
Introduction
The design and implementation of a comparability exercise in biosimilar development represent the fundamental benchmark for demonstrating molecular similarity between a proposed biosimilar and its reference biologic. Within the current regulatory framework, analytical characterization has evolved beyond its conventional quality control function and now serves as the central pillar of the biosimilarity dossier. It plays a decisive role in determining both the need for and the extent of subsequent clinical investigations. Since slight differences in quality attributes may result from variations in cell lines, manufacturing scales, and bioprocessing conditions, proving a high level of similarity requires a comprehensive, side-by-side structural and functional assessment. For biopharmaceutical developers, the analytical similarity package is the most influential factor affecting regulatory approval, intellectual property strategies, and overall development success. By applying advanced physical, chemical, and biological characterization techniques, highly specialized contract research organizations such as ResolveMass Laboratories Inc. help sponsors generate detailed, high-resolution molecular profiles that meet the rigorous regulatory expectations established by global health authorities.
Learn more about our comprehensive biosimilar characterization services.
Article Summary:
- A comparability exercise is the foundation of biosimilar development, demonstrating that a proposed biosimilar is highly similar to the reference biologic through comprehensive structural, physicochemical, and functional analyses rather than identical manufacturing processes.
- Successful study design begins with defining a Quality Target Product Profile (QTPP), selecting representative reference product lots, and accounting for natural manufacturing variability, process changes, and appropriate statistical acceptance criteria.
- Advanced analytical technologies—including LC-MS, peptide mapping, hydrogen-deuterium exchange mass spectrometry (HDX-MS), and orthogonal characterization methods—are used to confirm primary sequence, higher-order structure, post-translational modifications, and other critical quality attributes.
- Statistical approaches such as equivalence testing, Quality Range (QR) analysis, multivariate modeling, and risk-based evaluation help determine whether observed differences remain within scientifically acceptable limits while supporting reliable regulatory decisions.
- Comparability strategies for advanced therapies, including gene and cell therapies, require specialized paired-study designs and tailored statistical methods to minimize donor-related biological variability and strengthen analytical confidence.
- Regulatory agencies increasingly recognize that robust analytical, functional, pharmacokinetic, and immunogenicity evidence can provide sufficient assurance of biosimilarity, reducing the need for large comparative clinical efficacy studies in many cases.
- As biosimilar development continues to shift toward an analytical-first approach, comprehensive molecular characterization, sound statistical interpretation, and well-documented comparability data have become essential for efficient regulatory approval, lower development costs, and faster patient access to biologic therapies.

Designing a Robust Comparability Exercise in Biosimilar Development
A well-designed comparability exercise in biosimilar development begins with the establishment of a comprehensive Quality Target Product Profile (QTPP), developed through extensive characterization of a representative collection of reference product lots. This foundational profile captures the inherent manufacturing variability associated with the innovator product and defines the statistical boundaries required to demonstrate biosimilarity.
Instead of attempting to achieve exact molecular identity, which is unattainable because biologics are produced using living systems, sponsors are expected to demonstrate that the proposed biosimilar is highly similar to the reference product despite the presence of minor differences in clinically inactive components. The objective is to match the distribution of critical quality attributes rather than a single fixed value, reflecting the natural lot-to-lot variability that regulatory agencies recognize as an intrinsic characteristic of originator biologics. The overall study design must also consider factors such as reference product sourcing, lot age, and any manufacturing process changes introduced by the innovator throughout the product lifecycle.
The table below summarizes recommended lot selection strategies, statistical considerations, and study design parameters according to the US FDA final guidance and the EMA reflection paper guidelines.
| Parameter / Parameter Class | US FDA Regulatory Expectations | EMA CHMP Methodological Toolset |
|---|---|---|
| Reference Lot Procurement | At least 10 reference product lots collected over multiple years to adequately capture manufacturing variability. | Flexible lot acquisition strategy focused on representing the natural variability and long-term manufacturing capability of the reference biologic. |
| Proposed Biosimilar Lots | A minimum of 6 to 10 lots manufactured using the intended commercial-scale production process. | Flexible approach in which both clinical and commercial manufacturing batches are assessed to demonstrate process consistency. |
| Statistical Comparison Goal | Demonstrate that the biosimilar’s quality attributes fall within the predefined Quality Range (QR). | Supports flexible comparisons of means and variances, including tolerance intervals, Bayesian statistical approaches, and distribution overlap analyses. |
| Handling Process Shifts | Both pre-shift and post-shift reference product ranges may be used to establish similarity when they represent the licensed reference product. | Encourages analytical and clinical justification for observed manufacturing shifts by directly correlating differences with sensitive biological assays. |
Ensure your study meets regulatory standards with our biosimilar comparability studies.
Advanced Mass Spectrometry and Physicochemical Characterization in a Comparability Exercise in Biosimilar Development
Analytical characterization performed during a comparability exercise in biosimilar development depends on multiple orthogonal analytical platforms to verify both primary molecular structure and higher-order structural integrity. These advanced analytical technologies generate the detailed molecular fingerprint required to reduce residual uncertainty before progressing to clinical evaluation.
Modern biomanufacturing processes may introduce subtle structural differences, particularly in post-translational modifications (PTMs), which have the potential to influence therapeutic performance or increase immunogenicity. Complex biologics, including monoclonal antibodies and fusion proteins, are especially susceptible to degradation pathways such as deamidation, oxidation, and glycosylation heterogeneity. As a result, comprehensive, high-resolution structural characterization is essential. To achieve the level of analytical sensitivity expected by both the FDA and the EMA, sponsors are required to employ multiple independent analytical methods to confirm every critical quality attribute.
Explore our expertise in glycosylation analysis of biosimilars
and forced degradation of biosimilars.
Primary Structure Verification and Peptide Mapping Protocols
Verification of the primary structure requires complete amino acid sequence mapping supported by high-resolution liquid chromatography-mass spectrometry (LC-MS) to detect low-abundance sequence variants and chemically modified residues. This orthogonal analytical strategy confirms that the primary amino acid sequence of the proposed biosimilar is identical to that of the reference biologic.
To verify the complete protein sequence, laboratories perform a comprehensive peptide mapping workflow that includes protein denaturation, reduction, alkylation, and enzymatic digestion. Standard digestion procedures employ highly specific proteolytic enzymes such as trypsin, LysC, GluC, or pepsin to generate consistent and reproducible peptide fragments from the therapeutic protein. These peptide fragments are subsequently separated using Ultra-Performance Liquid Chromatography (UPLC) columns, including those manufactured with bio-inert titanium hardware to minimize metal adsorption of acidic deamidated species, before undergoing analysis with high-resolution tandem mass spectrometry.
This analytical strategy offers exceptional sensitivity, allowing the identification of codon-specific mistranslation events during protein biosynthesis. One example is the misincorporation of asparagine at serine residues, which produces a +27 Da mass shift and can be detected at levels ranging from 0.01% to 0.2%. In addition, peptide mapping delivers site-specific characterization of critical post-translational modifications (PTMs), including deamidation. Deamidation occurring within the complementarity-determining regions (CDRs) has the potential to interfere with antibody-antigen binding and is therefore regarded as a high-risk critical quality attribute. In contrast, deamidation located in the Fc region generally does not influence Fcγ or FcRn receptor binding and is considered a low-risk molecular variant.
We utilize a robust proteomics approach for biosimilars to ensure sequence accuracy.
Conformational Characterization and Higher-Order Structure (HOS) Profiling
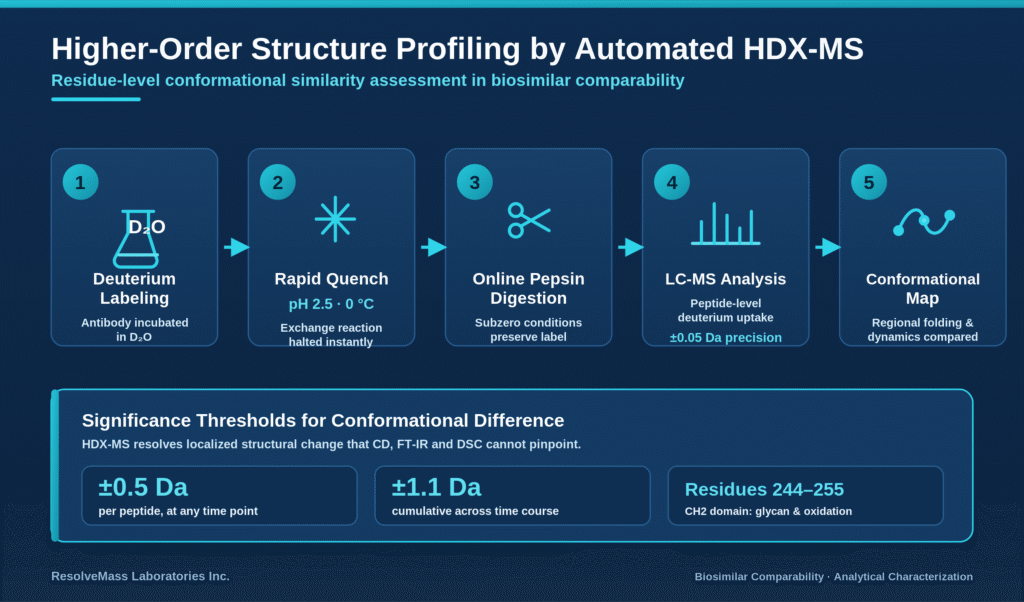
Conformational similarity assessment and higher-order structural profiling rely on automated hydrogen-deuterium exchange mass spectrometry (HDX-MS) to evaluate regional protein folding patterns and molecular dynamics under physiologically relevant conditions. By providing structural information at the residue level, this technique overcomes the spatial limitations associated with conventional global biophysical assays.
Unlike global analytical methods such as Fourier-Transform Infrared (FT-IR) spectroscopy and Circular Dichroism (CD) spectroscopy, which provide only an overall representation of secondary structural elements without identifying their precise locations, HDX-MS delivers localized insights into protein conformation and dynamic behavior. Within an automated HDX-MS workflow, the therapeutic antibody is incubated in deuterated water (D₂O) under carefully controlled temperature conditions. During this process, backbone amide hydrogens exchange with deuterium at rates determined by solvent accessibility and local hydrogen-bonding interactions, thereby generating a detailed map of the protein’s native three-dimensional structural dynamics.
The exchange reaction is rapidly terminated by reducing the pH to 2.5 while simultaneously lowering the temperature to 0 °C. This is followed by rapid online pepsin digestion and liquid chromatography-mass spectrometry (LC-MS) analysis performed under subzero temperature conditions. Highly automated and reproducible HDX-MS platforms routinely achieve a standard deviation of ±0.05 Da in deuterium uptake measurements. A mass difference (Δm) of ±0.5 Da for an individual peptide at any measured time point, or a cumulative difference of ±1.1 Da across the complete experimental time course, is recognized as the accepted threshold for identifying a statistically significant conformational difference.
For example, amino acid residues 244-255 located at the N-terminus of the CH2 domain have been shown to be particularly sensitive to changes in glycosylation profiles and methionine oxidation. Even subtle variations in this region, approximately 0.2 deuteron per peptide, may indicate localized changes in glycan composition or oxidation status. This demonstrates the value of HDX-MS in detecting localized conformational differences that remain undetected when using traditional global biophysical characterization methods.
| Analytical Attribute / Structural Level | Primary Characterization Platform | Supplementary / Orthogonal Technique | Software & System Controls |
|---|---|---|---|
| Intact Mass & Subunits | Non-reduced / Reduced RPLC-MS | Size-Exclusion Chromatography (SEC-MS) | UNIFI Software with MaxEnt1 Deconvolution algorithm |
| Primary Sequence Verification | UPLC-MS/MS Peptide Mapping | N/C-terminal sequencing, Amino acid analysis | Mascot, BioPharma Finder |
| Higher-Order Structure (HOS) | Automated HDX-MS | Far/Near-UV CD, FT-IR, DSC | DynamX HDX Data Analysis Software, HDExaminer |
| Charge Variant Heterogeneity | Weak Cation-Exchange (WCX-MS) | Capillary Isoelectric Focusing (cIEF) | BioResolve SCX columns, Empower software |
Statistical Frameworks and Data Interpretation in a Comparability Exercise in Biosimilar Development
Data interpretation within a comparability exercise in biosimilar development relies on risk-based statistical methodologies to assess analytical similarity while effectively controlling both Type I (patient) risk and Type II (sponsor) risk. These statistical approaches encompass rigorous hypothesis testing, multivariate clone screening techniques, and quality range calculations designed to support comprehensive similarity assessments.
To meet regulatory expectations, biosimilar developers must establish statistical acceptance criteria that account for both the clinical importance of individual quality attributes and the inherent mathematical limitations associated with relatively small sample sizes. Although the FDA’s original three-tier statistical framework historically served as the primary model for similarity assessment, current analytical data interpretation has progressed toward more flexible and scientifically robust statistical methodologies.
Learn more about our charge variant analysis in biosimilars
and impurity profiling of biosimilars.
Equivalence Testing and Margin Calculations
Equivalence testing is applied to high-risk critical quality attributes to confirm that the mean difference between the proposed biosimilar and the reference product remains within a predefined similarity margin. This evaluation is performed using the Two One-Sided Test (TOST) methodology, with the equivalence margin typically established as ±1.5 times the standard deviation of the reference biologic.
The standard FDA Tier 1 equivalence testing framework applies a fixed equivalence margin of ±1.5 σR, providing approximately 87% statistical power when analyses are conducted using 10 biosimilar lots and 10 reference product lots, assuming a true mean difference of 0.125 σR. However, replacing the unknown true standard deviation (σR) with a sample-derived estimate (σ̂R) does not adequately account for the statistical uncertainty associated with estimating variability from a limited number of manufacturing lots. In addition, when multiple analytical measurements are obtained from each reference lot, the standard FDA estimator may produce a biased estimate of the true reference standard deviation (σR). To overcome this limitation, Wang and Chow proposed alternative statistical methodologies based on nested variance estimators, enabling a more accurate representation of true lot-to-lot variability.
To provide greater flexibility than the fixed 1.5 σR equivalence margin, alternative statistical models permit the use of adjustable margins ranging from (1.575 σR, 2.025 σR) through application of a flexible index, denoted as f. For example, selecting a value of f = 1.25 expands the acceptance margin to ±1.875 σ̂R while maintaining the Type I error rate at the α = 5% significance level and providing more than 90% statistical power for demonstrating equivalence across varying standard deviation ratios. In situations involving multiple reference products, such as simultaneous comparisons with US-licensed and EU-authorized reference biologics, direct pairwise comparisons may generate inconsistent equivalence margins because the reference products possess different standard deviations. Under these circumstances, developers commonly employ Multiplicity-Adjusted Two One-Sided Tests (MATOST) or simultaneous confidence interval methods based on fiducial inference to establish directly comparable equivalence margins using a single primary reference product.
We provide specialized aggregation analysis in biosimilars
and precise insulin biosimilar characterization to support your development program.
Quality Range (QR) Approaches and Handling Innovator Process Shifts
The Quality Range (QR) approach is applied to evaluate moderate-risk quality attributes by confirming that a high proportion of biosimilar batch results fall within an acceptable range derived from the mean and standard deviation of the reference product. Under current regulatory expectations, this quality range is expressed as (μR ± XσR), where a multiplier between 2 and 3 is typically selected according to the criticality of the individual quality attribute.
According to the FDA’s September 2025 final guidance, the Quality Range methodology is recommended as the primary quantitative approach for comparative analytical data evaluation. Under this framework, analytical similarity is demonstrated when an adequate proportion of individual biosimilar lot values, typically 90%, fall within the Quality Range established from the reference product lots. To minimize the impact of small sample sizes or analytical method variability on the calculated Quality Range boundaries, developers may apply Quality Range Maximum Likelihood (QRML) estimation. This statistical approach estimates total variance by calculating the square root of the combined between-batch variance and within-batch variance, ensuring that analytical measurement error neither artificially expands nor narrows the acceptable Quality Range.
If the innovator biologic undergoes an authorized manufacturing process modification during the biosimilar development program, measurable shifts may occur in specific quality attribute profiles. Regulatory guidance recognizes that quality ranges established both before and after such an approved process change accurately represent the licensed reference product. Consequently, biosimilar developers may rely on either quality range when supporting the comparability exercise, since both manufacturing states correspond to clinically approved versions of the same reference biologic.
Ensure your data meets rigorous benchmarks through expert biosimilar characterization using mass spectrometry.
Statistical Considerations for Gene and Cellular Therapies
Comparability exercises designed for gene and cellular therapies should incorporate split-source paired study designs to effectively control the substantial donor-to-donor biological variability that is characteristic of these products. This paired analytical framework enhances statistical power while ensuring that any observed quality differences are attributable to manufacturing performance rather than inherent genetic variation among donors.
In autologous cell therapies, including CAR-T products, every manufacturing batch originates from a different patient or donor, creating considerable baseline variability in cellular composition, genetic background, and prior treatment history. As a result, conventional population-based equivalence testing and Quality Range methodologies are not statistically appropriate for these products. To minimize the influence of donor-specific biological variability, the FDA’s 2024 guidance recommends implementing a split-source study design. Under this approach, cellular source material obtained from a single donor is divided into two equal portions, with one portion processed using the pre-change manufacturing process and the other using the post-change process. This paired design effectively controls for donor-related biological differences, reduces batch-to-batch variability, and substantially improves the statistical power of the comparability assessment, even when working with highly limited sample sizes, such as N = 3. For gene therapies, historical manufacturing and analytical assay data may also be utilized to characterize analytical variability and establish the Equivalence Acceptance Criteria (EAC).
Achieve precise analytical alignment and prove biosimilarity using LC-MS to meet regulatory expectations.
Multivariate Clone Screening and Similarity Modeling
Multivariate analytical approaches employ advanced software algorithms to evaluate multiple correlated quality attributes simultaneously during the early stages of clone screening. By transforming high-dimensional analytical datasets into simplified similarity metrics, these methods enable developers to identify the most suitable production clones while minimizing analytical workload.
During the initial phases of biosimilar development, including strain selection and early process optimization, conducting independent inferential statistical analyses for numerous Critical Quality Attributes (CQAs) is both mathematically challenging and resource intensive. Software platforms such as Werum PAS-X Savvy apply Principal Component Analysis (PCA) together with multivariate statistical modeling to compare candidate production clones against the established reference product population. These analytical models generate multivariate similarity scores and orthogonal distance measurements while defining a 97.5% tolerance interval. Candidate batches that fall within this predefined region are considered highly similar to the reference product. Contribution plots are subsequently generated to identify the specific variables, including endotoxin concentration, protein titer, or individual charge variants, that contribute most significantly to any observed analytical differences. This information enables developers to make informed, data-driven manufacturing and process optimization decisions early in product development.
Optimize your early-stage development with intact mass analysis of biosimilars
and comprehensive peptide mapping in biosimilars.
Regulatory Expectations and the Evolving Clinical Efficacy Waiver Landscape
Current regulatory expectations allow biosimilar sponsors to request the waiver of comparative clinical efficacy studies when they can demonstrate highly similar analytical, functional, pharmacokinetic, and immunogenicity profiles. This evolving regulatory paradigm recognizes that modern physicochemical and biological analytical methods are often more sensitive than traditional clinical endpoints for detecting meaningful molecular differences.
The regulatory framework governing biosimilar development reached a significant milestone during late 2025. For more than fifteen years, regulatory agencies generally considered large Phase III comparative clinical efficacy studies (CES) essential for confirming therapeutic equivalence and resolving any remaining uncertainty regarding biosimilarity. However, cumulative evidence from more than 70 approved biosimilars demonstrated that no biosimilar possessing a robust analytical similarity package and pharmacokinetic (PK) similarity profile had failed a comparative clinical efficacy study. These findings confirmed that Phase III clinical trials primarily serve a confirmatory function rather than acting as sensitive tools for detecting molecular differences.
As a result, the FDA released its October 2025 draft guidance, Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Updated Recommendations for Assessing the Need for Comparative Efficacy Studies. This guidance shifts the role of comparative clinical studies from a routine regulatory requirement to a conditional scientific tool that is recommended only when residual uncertainty remains following comprehensive analytical and functional characterization. Considering that comparative clinical efficacy studies typically extend development timelines by approximately 1 to 3 years while adding development costs of nearly 24 million, the ability to waive these studies substantially reduces both development time and financial barriers to market entry.
The cornerstone of this regulatory flexibility is the submission of a highly persuasive analytical similarity package. When developers successfully demonstrate equivalent primary structure, comparable conformational dynamics through HDX-MS, similar distributions of post-translational modifications (PTMs), and matching biological activity across all relevant mechanisms of action, an appropriately designed human pharmacokinetic similarity study together with a tiered clinical immunogenicity assessment is generally considered sufficient to support regulatory approval.
Within this streamlined regulatory framework, every aspect of the analytical comparability program must be comprehensively documented in the Chemistry, Manufacturing, and Controls (CMC) section of the 351(k) Biologics License Application (BLA). Sponsors are expected to justify the selection of similarity acceptance criteria, reference product lots, and statistical methodologies. Any observed analytical differences must be thoroughly investigated using appropriate orthogonal analytical techniques and demonstrated to have no clinically meaningful impact, thereby ensuring that both patient safety and therapeutic efficacy remain fully protected.
A strong dossier depends on identifying post-translational modifications (PTMs) in biosimilars to ensure total structural similarity.
Conclusion
Conducting a scientifically rigorous comparability exercise in biosimilar development represents one of the most important scientific and strategic milestones for achieving efficient, cost-effective regulatory approval. By integrating advanced physicochemical characterization with robust statistical methodologies, sponsors can develop a comprehensive “totality of the evidence” package that supports clinical trial waivers while demonstrating therapeutic equivalence with confidence.
Biosimilar development has now firmly transitioned toward an analytical-first model, where high-resolution structural and functional characterization forms the basis for both regulatory and clinical success. Advanced analytical technologies, including automated peptide mapping and HDX-MS, provide the residue-level molecular resolution required to comprehensively characterize complex biologics, systematically reduce residual uncertainty, and demonstrate similarity within the natural variability of the reference product. As regulatory authorities such as the FDA and EMA continue to modernize approval pathways by reducing the reliance on unnecessary Phase III comparative clinical studies, the importance of partnering with an experienced analytical laboratory continues to grow.
ResolveMass Laboratories Inc. remains at the forefront of this evolving analytical landscape by delivering specialized mass spectrometry workflows, advanced biophysical characterization platforms, and regulatory expertise needed to design and execute fully compliant analytical similarity programs. Biopharmaceutical sponsors seeking to accelerate product development through state-of-the-art mass spectrometry solutions and customized statistical quality range strategies can connect with the technical and regulatory consulting team through the ResolveMass Laboratories Inc. Contact Page.
Frequently Asked Questions
The standard deviation (σR) of the reference product is a key factor in defining the acceptance limits for Tier 1 equivalence testing. A larger σR results in a broader equivalence margin, while a smaller σR produces a narrower acceptance range. If the reference variability is estimated from too few manufacturing lots, the calculated margin may not accurately represent the true product variability. This can increase the likelihood of statistical errors and affect the reliability of the biosimilarity assessment.
A flexible margin index (f) enables developers to adjust the equivalence margin based on the observed variability of the reference biologic rather than relying solely on a fixed statistical threshold. This approach improves the robustness of similarity testing, particularly when only a limited number of reference lots are available. It helps maintain appropriate control of Type I error while preserving high statistical power. As a result, the equivalence assessment becomes more representative of real-world manufacturing variability.
The expression system selected for manufacturing can influence several critical quality attributes, particularly post-translational modifications such as glycosylation patterns and charge variants. Although a biosimilar is not required to use the same host cell line as the reference product, any structural differences introduced by an alternative expression system must be carefully evaluated. Comprehensive analytical characterization and functional bioassays are necessary to demonstrate that these variations do not affect biological activity, safety, or immunogenicity.
A split-source design is a paired experimental approach commonly applied in autologous cell therapy comparability studies. In this design, cellular material obtained from a single donor is divided into two equal portions, with each portion processed using different manufacturing conditions. This strategy minimizes the impact of donor-specific biological variability and allows developers to evaluate manufacturing changes with greater statistical confidence. It is particularly valuable when only a small number of donor samples are available.
When a biosimilar is compared with both US-licensed and EU-authorized reference products, differences in variability between the reference products can complicate standard statistical comparisons. Multiplicity-adjusted statistical methods, including Multiplicity-Adjusted Two One-Sided Tests (MATOST), help address this challenge by maintaining consistent statistical confidence across multiple comparisons. These approaches improve the validity of similarity conclusions while supporting regulatory submissions across different global markets.
In HDX-MS analysis, the hydrogen-deuterium exchange reaction is rapidly stopped by lowering the pH to approximately 2.5 and reducing the temperature to 0 °C. These quench conditions minimize back-exchange and preserve the incorporated deuterium labels throughout sample preparation. The stabilized samples can then undergo rapid pepsin digestion followed by LC-MS analysis. This workflow ensures accurate characterization of protein conformation and higher-order structure.
Deamidation occurring within the complementarity-determining regions (CDRs) may directly influence antigen binding because these regions are responsible for target recognition. Consequently, CDR deamidation is considered a high-risk critical quality attribute that requires close analytical monitoring. In contrast, deamidation within the Fc region generally has little or no effect on Fcγ or FcRn receptor interactions. For this reason, Fc-region deamidation is usually classified as a lower-risk structural variation.
Quality Range Maximum Likelihood (QRML) estimation is used to establish statistically reliable quality range limits by incorporating both manufacturing batch variability and analytical method variability into the calculation. This approach reduces the influence of measurement uncertainty when defining similarity boundaries. As a result, QRML produces more accurate and stable acceptance ranges for moderate-risk quality attributes. It provides greater confidence in analytical similarity assessments across multiple production batches.
Yes. Regulatory agencies may approve a biosimilar with a different formulation, excipient composition, or delivery device if comprehensive evidence demonstrates that these differences do not negatively affect product quality or clinical performance. Developers must establish comparable stability, compatibility, biological activity, and safety through appropriate analytical and functional studies. Additional usability and device performance evaluations may also be required before approval.
Reference:
- Srinivasan, S., Kumar, R., & Kumar, S. (2022). Analytical similarity assessment of biosimilars: Global regulatory landscape, recent studies and major advancements in orthogonal platforms. Frontiers in Bioengineering and Biotechnology, 10, Article 832059. https://doi.org/10.3389/fbioe.2022.832059
- Curigliano, G., O’Connor, D. P., Rosenberg, J. A., & Jacobs, I. (2016). Biosimilars: Extrapolation for oncology. Critical Reviews in Oncology/Hematology, 104, 131–137. https://doi.org/10.1016/j.critrevonc.2016.06.002
- Webster, C. J., Woollett, G. R., & Thorpe, R. (2021). Comparability of biologics: Global principles, evidentiary consistency and unrealized reliance. BioDrugs, 35(4), 351–370. https://doi.org/10.1007/s40259-021-00490-0
- U.S. Food and Drug Administration. (2015). Scientific considerations in demonstrating biosimilarity to a reference product: Guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/media/101924/download
- Oliva, A., & Llabrés, M. (2021). New quality-range-setting method based on between- and within-batch variability for biosimilarity assessment. Pharmaceuticals, 14(6), 527. https://doi.org/10.3390/ph14060527
- U.S. Food and Drug Administration. (2025). Scientific considerations in demonstrating biosimilarity to a reference product: Updated recommendations for assessing the need for comparative efficacy studies: Draft guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/media/189366/download
- European Medicines Agency. (2021). Reflection paper on statistical methodology for the comparative assessment of quality attributes in drug development (EMA/CHMP/138502/2017). https://www.ema.europa.eu/en/statistical-methodology-comparative-assessment-quality-attributes-drug-development-scientific-guideline

