
Assessing the chemical safety of human drug products requires a thorough understanding of potential mutagenic and carcinogenic impurities that fall within the cohort of concern described under the International Council for Harmonisation (ICH) M7 guideline. For professionals working in quality assurance, regulatory affairs, and chemistry, manufacturing, and controls (CMC), one critical question frequently arises: Do All Drugs Need Nitrosamine Risk Assessment?
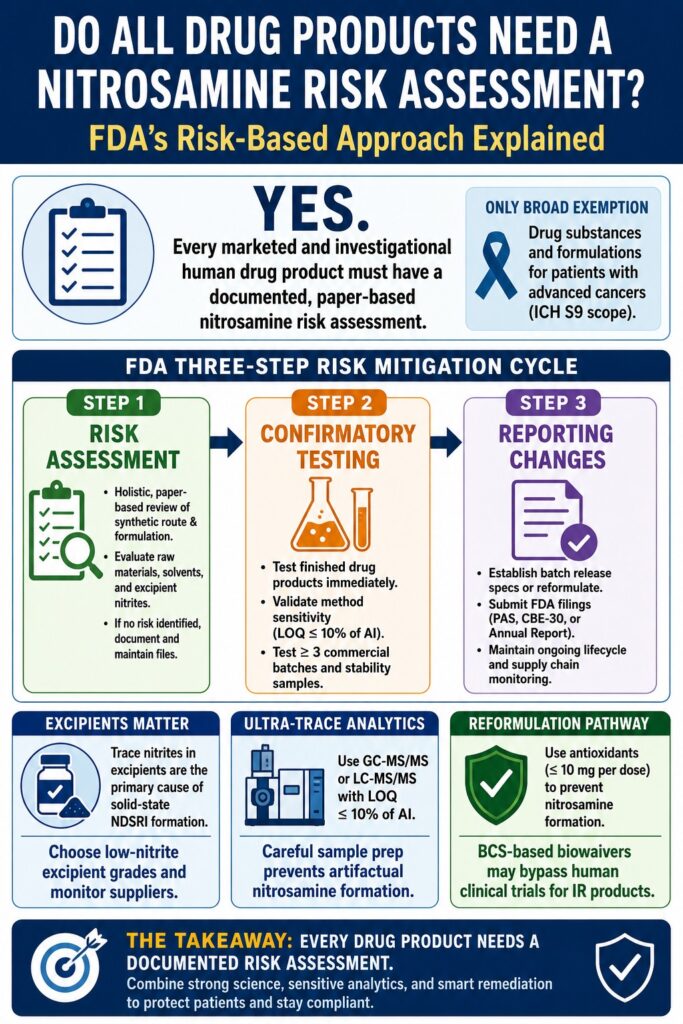
The answer is yes. Under the FDA’s comprehensive risk-based framework, every approved and marketed human drug product, as well as all products under clinical development through Investigational New Drug (IND) applications, must undergo a formal, documented, paper-based nitrosamine risk evaluation. Although laboratory testing is only required when a potential chemical vulnerability has been identified, a scientifically justified and well-documented risk assessment is a mandatory regulatory expectation for all applicable drug products.
Explore a foundational breakdown of these mutagenic impurities and why regulatory agencies prioritize them by reviewing our introductory guide: What are Nitrosamines?
Article Summary:
- All approved and investigational human drug products require a documented nitrosamine risk assessment under the FDA’s risk-based framework, even when laboratory testing is not initially necessary.
- Global regulatory agencies, including the FDA, EMA, Health Canada, and TGA, have aligned expectations that manufacturers maintain scientifically justified assessments across their entire product portfolios.
- Nitrosamine impurities are classified into small-molecule nitrosamines and NDSRIs, both of which can pose mutagenic and carcinogenic risks, though they differ in origin, structure, and control strategies.
- The FDA’s three-step mitigation process includes risk assessment, confirmatory testing, and corrective action, ensuring potential nitrosamine risks are identified, verified, and effectively managed.
- Trace nitrite impurities in pharmaceutical excipients are a major contributor to NDSRI formation, making supplier qualification and excipient selection critical components of risk reduction programs.
- Sensitive analytical techniques such as LC-MS/MS, GC-MS/MS, and HRMS are essential for detecting nitrosamines at extremely low levels while minimizing false-positive results during sample preparation.
- Remediation strategies, including formulation changes and antioxidant-based nitrite scavengers, can help reduce nitrosamine formation, while FDA biowaiver pathways may simplify compliance for certain reformulated immediate-release products.
Regulatory Boundary: Do All Drugs Need Nitrosamine Risk Assessment?
According to the FDA’s current risk-based guidance, all human drug products that are either commercially marketed or under development must undergo a formal, paper-based nitrosamine risk assessment. The only broad clinical exemption applies to drug substances and formulations intended for patients with advanced cancers, as defined within the scope of the ICH S9 guideline.
The worldwide regulatory response to nitrosamine contamination has continued to evolve since the 2018 detection of N-nitrosodimethylamine (NDMA) in the angiotensin II receptor blocker (ARB) valsartan. Since that time, the FDA’s Center for Drug Evaluation and Research (CDER) has made it clear that marketing authorization holders are responsible for maintaining documented evidence of risk evaluations throughout their product portfolios, including:
- Chemically synthesized active pharmaceutical ingredients (APIs) and synthetic intermediates.
- Semi-synthetic drug substances and fermentation-derived products.
- Biological products that contain chemically synthesized fragments or utilize nitrosating reagents during manufacturing.
- Over-the-counter (OTC) monograph drug products.
- All investigational human drug formulations undergoing clinical development through IND applications.
Regulatory agencies around the world, including the FDA, the European Medicines Agency (EMA), Health Canada, and Australia’s Therapeutic Goods Administration (TGA), have largely aligned their expectations regarding nitrosamine risk assessments. Although biological products generally present a lower baseline risk because of highly purified water systems and purification techniques based on molecular size, they are not exempt from the assessment process. For example, antibody-drug conjugates (ADCs) that employ synthetic drug linkers have emerged as a recognized source of vulnerability for Nitrosamine Drug Linker-Related Impurities (NDLRIs), requiring proactive chemical evaluation.
In addition, historical findings have demonstrated that theoretical risk assessments alone may not always predict real-world outcomes. In several instances, Official Medicines Control Laboratories (OMCLs) identified complex nitrosamines in finished pharmaceutical products despite manufacturing reviews indicating no apparent risk. These findings reinforce the regulatory expectation that every product within a portfolio must be supported by a complete, documented, paper-based risk assessment capable of withstanding regulatory scrutiny and audits.
Review the specific guidelines, mutagenic classifications, and compliance updates governing impurity testing: Genotoxic Impurity Testing & ICH M7 Nitrosamines
Assess risk profiles, safety metrics, and distinct vulnerabilities associated with macromolecular therapeutics: Nitrosamine Impurities in Biologics
Chemistry of Concern: Small-Molecule Nitrosamines versus NDSRIs
The FDA differentiates between small-molecule nitrosamines, which are structurally unrelated to the API, and Nitrosamine Drug Substance-Related Impurities (NDSRIs), which share structural characteristics with the active pharmaceutical ingredient. Both categories are considered highly potent mutagenic compounds capable of damaging DNA after metabolic activation. Through the formation of electrophilic diazonium ions, these compounds can alkylate and methylate DNA bases, potentially leading to genomic instability and carcinogenesis.
| Parameter | Small-Molecule Nitrosamines | Nitrosamine Drug Substance-Related Impurities (NDSRIs) |
|---|---|---|
| Structural Profile | Low-molecular-weight, volatile compounds unrelated to the API structure. | Large, complex, non-volatile compounds directly derived from the API molecular structure. |
| Common Examples | NDMA, NDEA, NMBA, NIPEA, NDIPA, NDBA, NMPA. | N-nitroso-duloxetine, N-nitroso-fluoxetine, N-nitroso-vortioxetine. |
| Primary Origin | Process-related reactions involving secondary or tertiary amine solvents such as DMF and NMP with nitrous acid during API synthesis. | Solid-state reactions between vulnerable amine groups within the API and trace nitrites present in excipients during formulation or storage. |
| Purging Capability | High; volatile compounds can often be removed through crystallization, distillation, washing, or other downstream purification processes. | Extremely limited; once formed in the finished dosage form during storage, they cannot be physically removed. |
| Default AI Limit | Controlled through compound-specific toxicological data, such as 96 ng/day for NDMA and 26.5 ng/day for NDEA. | Calculated using structural activating and deactivating factors under the CPCA framework. |
For complex NDSRIs lacking compound-specific carcinogenicity data, the FDA, EMA, and Health Canada support the use of the Carcinogenic Potency Categorization Approach (CPCA). This structural toxicology methodology evaluates chemical characteristics such as the number of hydrogen atoms located on carbon atoms alpha to the nitroso group (α-hydrogen feature scores) and the presence of electronic deactivating groups to predict carcinogenic potency.
The CPCA assigns NDSRIs into one of five potency categories, which determine the corresponding acceptable intake (AI) limits.
│
▼
[Assess α-Hydrogen Configuration]
(0,0 / 0,1 / 0,2 / 0,3 / 1,1 / 1,2 / 1,3 / 2,2 / 2,3 / 3,3)
│
▼
(Carboxylic acids, Steric hindrance, Ring geometry)
│
▼
│
▼
[Assign CPCA Category (1 through 5)]
(Limits: 26.5 ng/day to 1500 ng/day)Structural deactivating features significantly reduce the likelihood of cytochrome P450-mediated metabolic activation, thereby lowering carcinogenic potential. For example, the presence of a carboxylic acid group anywhere within the molecule contributes +3 points to the structural score, while an N-nitroso group contained within a five- or six-membered ring contributes +2 points.
In contrast, molecules lacking deactivating features and containing highly reactive α-hydrogens, such as those exhibiting a 3,3 configuration, are assigned to Category 1, resulting in the most stringent acceptable intake limit of 26.5 ng/day.
Trace the mechanisms of secondary and tertiary amine interactions during active pharmaceutical ingredient synthesis: Nitrosamine Formation Pathways in API Synthesis
The FDA Three-Step Risk Mitigation Cycle
The FDA applies a structured three-step mitigation framework that requires manufacturers to identify potential risks, analytically confirm vulnerabilities, and implement sustainable control measures. This lifecycle-based approach is designed to ensure that all potential pathways for nitrosamine introduction or formation are systematically evaluated and controlled.
┌────────────────────────────────────────────────────────────────────────┐
│ STEP 1: RISK ASSESSMENT │
│ • Conduct holistic, paper-based synthetic route & formulation review. │
│ • Evaluate raw materials, solvent recovery, and excipient nitrites. │
│ • If no risk is identified, document internally and maintain files. │
└────────────────────────────────────────────────────────────────────────┘
│
Vulnerability Identified
▼
┌────────────────────────────────────────────────────────────────────────┐
│ STEP 2: CONFIRMATORY TESTING │
│ • Immediately trigger analytical testing of finished drug products. │
│ • Validate method sensitivity (LOQ ≤ 10% of AI target). │
│ • Test a minimum of 3 commercial batches and stability samples. │
└────────────────────────────────────────────────────────────────────────┘
│
Impurity Detected Above Limit
▼
┌────────────────────────────────────────────────────────────────────────┐
│ STEP 3: REPORTING CHANGES │
│ • Establish routine batch release specifications or reformulate API. │
│ • Submit variation filings to FDA (PAS, CBE-30, or Annual Report). │
│ • Maintain ongoing lifecycle and supply chain monitoring. │
└────────────────────────────────────────────────────────────────────────┘The execution of this three-step process must be thoroughly documented within the pharmaceutical quality system to ensure readiness for regulatory inspections and audits.
Step 1: Risk Assessment
The first phase involves a comprehensive paper-based review of both active and inactive ingredients. Manufacturers must systematically assess raw materials, vendor-supplied components, synthetic routes, processing aids, catalysts, recycled solvents, process water, packaging materials, and other environmental factors that may contribute to nitrosamine formation.
If no credible risk of nitrosamine formation or cross-contamination is identified during Step 1, physical laboratory testing is not required. However, a detailed and scientifically justified assessment report must be prepared and maintained on-site.
The FDA does not consider blanket statements such as “no amines are present” to constitute an adequate risk assessment.
Examine the impact of the updated structural guidelines and step-by-step risk workflows under the latest ICH modifications: Impact of ICH M7(R2) Updates on Nitrosamine Risk Assessment
Step 2: Confirmatory Testing
If Step 1 identifies a potential chemical vulnerability, manufacturers must proceed immediately to confirmatory analytical testing. The FDA expects testing to include at least three representative commercial batches of the finished drug product.
In addition, sponsors are encouraged to evaluate samples from both accelerated and long-term stability programs to determine whether nitrosamines may accumulate throughout the product’s shelf life.
All analytical methods employed must be appropriately validated and capable of achieving the sensitivity necessary to detect nitrosamines at trace levels.
Step 3: Reporting Changes
When confirmatory testing identifies nitrosamines above established action limits, manufacturers must move directly to Step 3. This phase involves implementing a permanent control strategy, such as process optimization, supplier qualification changes, raw material replacement, or product reformulation, followed by submission of the necessary regulatory filings.
Depending on the nature and impact of the changes, submissions may be filed as a Prior Approval Supplement (PAS), a Changes Being Effected in 30 Days (CBE-30) supplement, or an Annual Report.
Although the original deadline for completing Step 3 marketing authorization updates for NDSRIs was August 1, 2025, the FDA revised its enforcement approach in June 2025. Under current enforcement policies, manufacturers engaged in active remediation efforts may submit detailed progress reports to the agency in order to maintain product availability and avoid import alerts or recalls, provided that meaningful good-faith efforts are being demonstrated.
In addition, while theoretical purge calculations and in silico modeling tools, including Lhasa’s Mirabilis software, may support an “Option 4” control strategy during synthetic route evaluation, the FDA frequently expects empirical testing of the finished drug product to supplement theoretical predictions.
Review the multi-year compliance landmarks and strategic milestones required to maintain market access: Nitrosamine Testing Timeline and Milestones
Excipient Vulnerabilities: The Impact of Trace Nitrite Impurities
Trace nitrite impurities found in commonly used pharmaceutical excipients represent the primary cause of solid-state NDSRI formation in finished drug products. Under acidic or elevated-temperature conditions, nitrite contaminants readily convert into nitrous acid (HNO₂) and reactive nitrosating nitrogen oxides (NOₓ), which can react directly with vulnerable secondary or tertiary amine-containing APIs.
│
(Acidic / Thermal Activation)
▼
[Nitrous Acid Formation]
│
(Dimerization & Dehydration)
▼
[Nitrous Anhydride (N2O3)]
│
(Reaction with Amine API)
▼Nitrite concentrations vary significantly among excipient categories, manufacturing batches, and raw material suppliers.
| Excipient Class | Common Brand Names | Median Nitrite Level (ppm) | Hazard Assessment for Vulnerable APIs |
| Microcrystalline Cellulose (MCC) | Pharmacel | <0.1 | Very low risk; minimal contribution of nitrosating agents. |
| Milled and Sieved Lactose | Pharmatose, Lactochem | <0.1 | Very low risk; highly compatible with sensitive APIs. |
| Direct Compression Lactose (Standard) | SuperTab | <0.1 | Very low risk; suitable for direct-compression formulations. |
| Direct Compression Lactose (Specialty) | SuperTab NZ | 0.7 | Moderate risk due to elevated trace nitrite and nitrate levels. |
| Sodium Starch Glycolate (SSG) | Primojel | <0.1 | Low risk despite elevated nitrate concentrations. |
| Croscarmellose Sodium (CCS) | Primellose | <0.1 | Low risk with minimal nitrite contribution. |
| Crospovidone (Superdisintegrant) | Polyvinylpolypyrrolidone | 0.9–1.2 | High risk; nitrite concentrations are approximately 9- to 12-fold higher than MCC and lactose. |
Because of this substantial variability, the FDA encourages manufacturers to establish supplier qualification programs that actively monitor nitrite levels in incoming excipient lots. Selecting low-nitrite excipient grades and replacing high-nitrite binders or disintegrants can significantly reduce the likelihood of NDSRI formation during storage.
Evaluate specific compound risks, testing protocols, and case studies for high-risk therapeutic types like adrenergic blockers: Nitrosamine Testing for Beta-Blockers
Analytical Demands: Instrumentation, Sensitivity, and Sample Preparation
Nitrosamine analytical methods must achieve parts-per-billion (ppb) sensitivity to accurately quantify trace mutagenic impurities within complex pharmaceutical formulations. The FDA expects laboratories to employ advanced chromatographic systems coupled with mass spectrometry, including Ultra-Performance Liquid Chromatography-Tandem Mass Spectrometry (UPLC-MS/MS) and Gas Chromatography-Tandem Mass Spectrometry (GC-MS/MS).
Sample preparation represents one of the most critical stages of trace nitrosamine analysis. Extracting nanogram-level impurities from highly concentrated pharmaceutical matrices requires careful method optimization to preserve analytical sensitivity while preventing artificial nitrosamine formation.
┌────────────────────────────────────────────────────────────────────────┐
│ WEIGHING & GRINDING │
│ • Grind tablets with a mortar and pestle to increase surface area. │
│ • Weigh samples using calibrated analytical balances. │
└────────────────────────────────────────────────────────────────────────┘
│
▼
┌────────────────────────────────────────────────────────────────────────┐
│ SOLVENT EXTRACTION │
│ • Add a target-specific solvent containing nitrite scavengers. │
│ • Vortex and mechanically agitate to ensure complete extraction. │
└────────────────────────────────────────────────────────────────────────┘
│
▼
┌────────────────────────────────────────────────────────────────────────┐
│ CENTRIFUGATION & FILTRATION │
│ • Separate analytes from solid residue through centrifugation. │
│ • Filter through inert membranes to protect chromatographic systems. │
└────────────────────────────────────────────────────────────────────────┘
│
▼
┌────────────────────────────────────────────────────────────────────────┐
│ HYPHENATED CHROMATOGRAPHIC ANALYSIS │
│ • Analyze using GC-MS/MS or LC-MS/MS platforms. │
│ • Verify that LOQ satisfies the required 10% AI threshold. │
└────────────────────────────────────────────────────────────────────────┘One of the most significant challenges during sample preparation is preventing artifactual nitrosamine formation. For example, excessive energy generated during aggressive sonication can accelerate nitrosation reactions between residual nitrites and amine-containing APIs, leading to false-positive results. To minimize this risk, analytical protocols should maintain low extraction temperatures and incorporate nitrite scavengers such as sulfamic acid into extraction solvents.
Furthermore, characterization of complex NDSRIs often requires High-Resolution Mass Spectrometry (HRMS) to distinguish target analytes from structurally similar APIs and matrix components that may cause chromatographic interference, ion suppression, or inaccurate quantification.
To support regulatory compliance, ResolveMass Laboratories Inc. employs validated GC-MS/MS and LC-MS/MS methodologies with limits of quantitation (LOQs) well below the required 10% acceptable intake threshold. This enables reliable detection and quantification of trace-level nitrosamine impurities while generating robust data suitable for regulatory submissions and audits.
Discover trace-level quantification strategies, instrument parameters, and testing methodologies for low-level detections: Ultra-Low Limit of Quantitation (LOQ) in Nitrosamine Testing
Read a direct performance review regarding sample injection styles, thermal volatility, and matrix separation options: Direct Injection vs. Headspace Techniques for Nitrosamines
Remediation Protocols: Reformulation and Bioequivalence Biowaivers
Reformulating drug products with trace nitrite scavengers is among the most effective strategies for mitigating solid-state NDSRI formation. To facilitate compliance efforts, the FDA’s revised September 2024 guidance introduced alternative bioequivalence (BE) pathways that may eliminate the need for traditional human clinical studies for certain reformulated immediate-release (IR) products.
│
▼
(Ascorbic acid, α-tocopherol, or propyl gallate ≤ 10 mg)
│
▼
│
┌────────────────────────┴────────────────────────┐
▼ ▼
(High API Water Solubility) (Low API Water Solubility)
│ │
▼ ▼
[Case-by-Case Evaluation]
(Multi-pH comparative profiles) (PBPK modeling or IVIVC)
│ │
▼ ▼
(Bypass human clinical trials) (Streamlined clinical data)The incorporation of approved antioxidants such as ascorbic acid, α-tocopherol, propyl gallate, or cysteine hydrochloride at levels of ≤10 mg per dose has demonstrated effectiveness in suppressing nitrosamine formation within solid-state formulations.
Under the FDA’s updated biowaiver framework, immediate-release products reformulated with these scavengers may qualify for alternative bioequivalence pathways based on the Biopharmaceutics Classification System (BCS).
BCS Class I and Class III
BCS Class I (High Solubility, High Permeability) and BCS Class III (High Solubility, Low Permeability) products may qualify for in vivo bioequivalence waivers because extensive FDA research has shown that small quantities of antioxidants (≤10 mg per dose) do not significantly affect API absorption or intestinal transporter activity.
Sponsors may obtain a biowaiver by submitting comparative multi-pH dissolution profiles demonstrating equivalent drug release characteristics before and after reformulation.
BCS Class II and Class IV
BCS Class II (Low Solubility, High Permeability) and BCS Class IV (Low Solubility, Low Permeability) products require more extensive evaluation because absorption and dissolution are highly sensitive to formulation changes.
In these cases, biowaiver eligibility is determined individually. Sponsors must provide comparative dissolution data across multiple pH conditions along with Physiologically Based Pharmacokinetic (PBPK) modeling and/or in vitro-in vivo correlation (IVIVC) evidence demonstrating that the formulation change does not adversely affect drug absorption.
This risk-based alternative pathway is not applicable to narrow therapeutic index (NTI) drugs and does not extend to sublingual, buccal, or orally disintegrating dosage forms. Nevertheless, for many immediate-release tablets and capsules, this framework substantially reduces both the cost and timeline associated with nitrosamine remediation.
View complete validation pipelines, tech-transfer frameworks, and assay optimization workflows: Nitrosamine Method Development and Validation Services
Scientific Synthesis and Strategic Action
Effective nitrosamine risk management requires an ongoing quality system that combines scientifically justified theoretical assessments with advanced analytical confirmation. Successfully navigating this increasingly complex regulatory landscape demands collaboration with experienced scientific partners capable of delivering validated trace-level analysis and formulation expertise.
Although the question, “Do All Drugs Need Nitrosamine Risk Assessment?” has a straightforward answer—yes, documented paper-based assessments are required for all applicable products—the greater commercial challenge lies in performing highly sensitive confirmatory testing and implementing successful remediation strategies without triggering costly clinical studies.
Learn how outsourcing to a specialized partner can help you streamline risk workflows and secure regulatory approval: Outsourcing Nitrosamine Testing to a CRO
For comprehensive nitrosamine risk assessments, expert regulatory consulting, advanced formulation support, and GMP-compliant confirmatory testing services, contact ResolveMass Laboratories Inc. at:
Frequently Asked Questions (FAQs)
No, analytical laboratory testing is not automatically required for every marketed drug product. The FDA’s risk-based framework begins with a comprehensive paper-based assessment to determine whether any credible pathway exists for nitrosamine formation or contamination. When this initial review shows no meaningful risk factors, additional laboratory testing may not be necessary. However, manufacturers must maintain detailed documentation that supports their conclusions and demonstrates compliance during regulatory inspections.
A drug substance may be considered a potential NDSRI precursor if it contains chemical groups that are susceptible to nitrosation reactions. These typically include secondary amines, tertiary amines that can be converted into secondary amines through metabolic or chemical processes, and certain amide-containing structures. When exposed to trace nitrosating agents, these functionalities may react and form nitrosamine drug substance-related impurities. Identifying these structural alerts is a key part of the initial risk assessment process.
The 10% Acceptable Intake (AI) threshold serves as an important trigger for additional quality control requirements. If confirmatory testing consistently detects a nitrosamine impurity above 10% of its established AI limit, manufacturers are generally expected to incorporate that impurity into routine release and stability specifications. This means ongoing monitoring becomes part of the product’s quality system. As a result, every commercial batch may require testing before being released for distribution.
The FDA has established compound-specific acceptable intake limits to minimize long-term patient exposure to highly potent nitrosamines. For N-nitrosodimethylamine (NDMA), the acceptable intake limit is 96 ng/day, while for N-nitrosodiethylamine (NDEA), the limit is 26.5 ng/day. These values are based on toxicological and carcinogenicity data used to estimate negligible lifetime cancer risk. Maintaining exposure below these limits is considered protective of patient safety.
No, over-the-counter (OTC) monograph drug products are not exempt from nitrosamine risk assessment requirements. Manufacturers of OTC products are expected to conduct the same structured Step 1 evaluations that apply to prescription medications. This includes reviewing raw materials, manufacturing processes, formulation components, and potential contamination pathways. Supporting documentation must be retained and made available for regulatory review when requested.
Physical purging can be an effective strategy for reducing or eliminating certain process-related small-molecule nitrosamines during API manufacturing. However, it cannot fully replace finished product testing when there is a risk of NDSRI formation. Unlike volatile nitrosamines that may be removed through purification steps, NDSRIs often develop later during formulation or storage. Therefore, final product testing may still be required to verify that nitrosamine levels remain within acceptable limits throughout the product lifecycle.
Trace nitrite concentrations can differ substantially among excipient manufacturers, production lots, and raw material sources. Even excipients that perform the same pharmaceutical function may contain significantly different nitrite levels depending on how they are produced. For example, studies have shown that crospovidone can contain nitrite concentrations many times higher than those found in microcrystalline cellulose or lactose. This variability highlights the importance of supplier qualification programs and routine raw material monitoring.
Advanced mass spectrometry-based analytical techniques are considered the industry standard for nitrosamine testing. Ultra-Performance Liquid Chromatography-Tandem Mass Spectrometry (UPLC-MS/MS) and Gas Chromatography-Tandem Mass Spectrometry (GC-MS/MS) are the most commonly used platforms because they provide exceptional sensitivity and selectivity. These technologies can accurately detect impurities at parts-per-billion levels within complex pharmaceutical matrices. Their performance makes them particularly suitable for regulatory compliance and confirmatory testing applications.
Yes, certain drug products intended for patients with advanced cancers may be exempt from specific nitrosamine acceptable intake recommendations. These exemptions fall within the scope of the ICH S9 guideline, which recognizes that the immediate therapeutic benefits of treatment may outweigh the theoretical long-term risks associated with low-level mutagenic impurities. While risk management principles still apply, regulatory expectations may differ for these specialized clinical settings. Decisions are generally made based on an overall benefit-risk assessment.
Reference:
- U.S. Food and Drug Administration. (2024, September). Control of nitrosamine impurities in human drugs: Guidance for industry (Revision 2). U.S. Department of Health and Human Services. https://www.fda.gov/media/187315/download
- U.S. Food and Drug Administration. (2021, November 9). Updates on possible mitigation strategies to reduce the risk of nitrosamine drug substance-related impurities in drug products. U.S. Department of Health and Human Services. https://www.fda.gov/drugs/drug-safety-and-availability/updates-possible-mitigation-strategies-reduce-risk-nitrosamine-drug-substance-related-impurities
- U.S. Food and Drug Administration. (2025). CDER nitrosamine impurity acceptable intake limits. U.S. Department of Health and Human Services. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/cder-nitrosamine-impurity-acceptable-intake-limits
- U.S. Food and Drug Administration. (2024). Determining recommended acceptable intake limits for N-nitrosamine impurities in pharmaceuticals: Development and application of the carcinogenic potency categorization approach. U.S. Department of Health and Human Services. https://www.fda.gov/drugs/spotlight-cder-science/determining-recommended-acceptable-intake-limits-n-nitrosamine-impurities-pharmaceuticals
- Teasdale, A. (2025). Guidelines for the assessment and control of mutagenic impurities in pharmaceuticals. Journal of Pharmaceutical Health Care and Sciences, 11, Article 1. https://pmc.ncbi.nlm.nih.gov/articles/PMC12723853/
- Health Canada. (2022, April 4). Nitrosamine impurities in medications: Guidance. Government of Canada. https://www.canada.ca/en/health-canada/services/drugs-health-products/compliance-enforcement/information-health-product/drugs/nitrosamine-impurities/medications-guidance.html
- Naiffer_Host. (2023, August 4). FDA – Recommended acceptable intake limits for NDSRIs guidance for industry [Online forum post]. Nitrosamines Exchange. https://nitrosamines.usp.org/t/fda-recommended-acceptable-intake-limits-for-ndsris-guidance-for-industry/7145

