 Testing vs. Drug-Device Compatibility Studies What Is the Difference")
Introduction:
E&L testing vs drug device compatibility studies comes down to one core distinction: E&L testing asks what chemicals migrate into a drug product and whether they are safe, while compatibility studies ask whether the drug and device continue to work together correctly over time. Both are essential for injectables, ophthalmics, inhalation products, biologics, prefilled syringes, autoinjectors, wearable injectors, and infusion systems, and both are typically expected in a single, risk-based regulatory submission.
Although the two study types are often mentioned in the same breath, they answer entirely different scientific questions and rely on different disciplines — analytical chemistry and toxicology for E&L, and formulation science and device engineering for compatibility. Together, they demonstrate product quality, patient safety, and long-term performance.
Why Are Both Studies Required?
Both studies reduce a different category of risk, and neither substitutes for the other.
E&L testing answers:
- Are harmful chemicals migrating into the drug product?
- What compounds are present, and at what concentration?
- Do those compounds pose a toxicological concern?
Drug-device compatibility studies answer:
- Does the drug remain stable inside the device?
- Does the delivery device perform correctly and consistently?
- Is dosing accurate across the shelf life?
- Are device materials altered by the formulation, or vice versa?
Regulators increasingly expect manufacturers to evaluate both chemical migration and functional compatibility before approving complex combination products, particularly as scrutiny around nitrosamine impurities in packaging systems and other high-risk leachables continues to grow.
Summary:
- E&L Testing vs Drug Device Compatibility Studies are complementary but fundamentally different evaluations required throughout pharmaceutical and combination product development.
- Extractables and leachables (E&L) testing identifies and quantifies chemicals that migrate from packaging, delivery devices, and manufacturing components into the drug product.
- Drug-device compatibility studies evaluate whether the drug formulation and delivery device function safely and effectively together across the product’s shelf life and clinical use.
- E&L testing is primarily a chemical safety and toxicology exercise; compatibility studies evaluate physical, mechanical, functional, and stability interactions.
- FDA, EMA, USP, ICH, and ISO frameworks increasingly expect both datasets for combination products such as prefilled syringes, autoinjectors, inhalers, and infusion systems.
- Early, integrated study design reduces regulatory delays, redundant testing, product redesigns, and commercialization risk.
- ResolveMass Laboratories Inc. designs and executes both E&L and compatibility programs using advanced mass spectrometry and regulatory-aligned study design.
1: What Is Extractables and Leachables (E&L) Testing?
Extractables and leachables testing identifies, characterizes, and quantifies chemicals that can migrate from packaging materials, manufacturing components, or medical devices into a pharmaceutical product. These chemicals may originate from:
- Plastic additives and plasticizers
- Antioxidants and stabilizers
- Colorants and catalysts
- Oligomers and residual monomers
- Processing aids and lubricants
The objective is to determine whether migrated compounds pose a toxicological risk to patients, an area covered in detail in our review of extractables and leachables thresholds and how safety concern thresholds are set.
Understanding Extractables
Extractables are compounds intentionally forced out of materials using aggressive, worst-case extraction conditions — organic solvents, acidic and basic solutions, elevated temperatures, and extended extraction durations. Selecting the right extraction media is a critical design decision; our guide to solvent selection for extractables studies outlines how solvent polarity and extraction conditions influence the completeness of the chemical inventory. Extractables studies establish the full universe of compounds that could potentially migrate during storage and use.
Understanding Leachables
Leachables are the subset of extractables that actually migrate into the pharmaceutical product during manufacturing, storage, transportation, or clinical use — and unlike extractables, they represent real patient exposure. Leachables studies are generally performed under real storage conditions, throughout shelf life, using the actual drug formulation, across multiple stability time points.
Common Sources of Extractables and Leachables
Potential sources include:
- Rubber stoppers and elastomeric closures
- Syringe plungers, prefilled syringes, and autoinjectors
- IV bags, infusion tubing, and filters
- Single-use bioprocess systems
- Plastic bottles and other plastic packaging components
- Ophthalmic drug product containers
- Nasal spray pumps, metered-dose inhalers, and dry powder inhalers
Because container closure systems vary widely in polymer chemistry and manufacturing process, E&L testing for container closure systems is generally scoped on a system-specific, risk-based basis rather than a one-size-fits-all protocol.
Analytical Techniques Used in E&L Testing
Comprehensive E&L programs typically combine multiple orthogonal analytical platforms to capture volatile, semi-volatile, non-volatile, and elemental species.
| Analytical Technique | Primary Purpose |
|---|---|
| LC-HRMS | Non-volatile organic compounds |
| LC-MS/MS | Targeted leachables |
| GC-MS/MS | Volatile and semi-volatile compounds |
| GC-HRMS | Unknown compound identification |
| ICP-MS | Elemental impurities and metals |
| FTIR | Polymer characterization |
| UV-Visible Spectroscopy | Certain extractable compounds |
| Headspace GC | Residual solvents and volatile species |
Choosing between platforms is rarely either/or — our comparison of GC-MS vs. LC-MS in extractables and leachables testing explains how the two techniques complement each other across a full E&L program, while ICP-MS in extractables and leachables testing covers elemental impurity detection specifically. Advanced high-resolution mass spectrometry enables confident identification of unknown leachables present at trace levels.
2: What Are Drug-Device Compatibility Studies?
Drug-device compatibility studies are designed to determine whether a pharmaceutical formulation and its delivery device remain safe, stable, and fully functional throughout the product’s intended shelf life and clinical use. Unlike Extractables and Leachables (E&L) testing, which focuses on chemical migration from packaging or device materials, compatibility studies evaluate the overall interaction between the drug product and the delivery system. These studies ensure that the formulation does not adversely affect the device and that the device continues to deliver the correct dose with consistent performance over time. They are particularly important for combination products such as prefilled syringes, autoinjectors, inhalers, infusion pumps, wearable injectors, and ophthalmic delivery systems. By assessing both pharmaceutical stability and device functionality, compatibility studies help demonstrate product quality, patient safety, and regulatory compliance throughout the product lifecycle.
What Do Drug-Device Compatibility Studies Evaluate?
Drug stability — One of the primary objectives is to confirm that the drug formulation remains chemically and physically stable while stored within the delivery device. Scientists evaluate whether the active pharmaceutical ingredient (API) maintains its potency, whether degradation products increase over time, and whether protein aggregation or precipitation occurs in biologics. Additional assessments include monitoring pH changes, color or appearance alterations, particulate formation, viscosity, and other critical quality attributes throughout real-time and accelerated stability studies.
Device functionality — Compatibility studies also verify that the delivery device performs consistently throughout its intended shelf life. Testing includes evaluating injection force, break-loose force, glide force, spray pattern, aerosol performance for inhalation products, dose delivery accuracy, residual volume, actuation consistency, and overall device reliability. These evaluations ensure that patients receive the intended dose accurately and safely during every use.
Material compatibility — Drug formulations may interact with materials used in medical devices, potentially affecting both the formulation and the device. Compatibility studies assess whether prolonged contact with the formulation causes swelling, degradation, extraction, adsorption, or other changes in elastomers, silicone oil lubricants, adhesives, plastic polymers, needle coatings, gaskets, or other device components. Identifying these interactions early helps prevent product failures and extends device reliability.
Mechanical integrity — Maintaining the structural integrity of the delivery system is essential for preserving sterility and ensuring proper performance. Mechanical evaluations include container closure integrity testing (CCIT), seal strength assessments, leakage testing, crack resistance, pressure resistance, stress testing, and inspection for component deformation or material fatigue. These tests verify that the device can withstand manufacturing, transportation, storage, and routine patient use without compromising product quality.
Long-term stability — Drug-device compatibility must be demonstrated throughout the entire product lifecycle rather than at a single time point. Therefore, studies are performed at initial release and continue under accelerated, intermediate, and long-term stability conditions according to ICH guidelines. Transportation simulation and shipping studies may also be conducted to evaluate the effects of vibration, temperature fluctuations, and handling on both the formulation and the delivery device. These long-term assessments provide confidence that the product will remain safe, effective, and fully functional until the end of its labeled shelf life.

3: Extractables and Leachables (E&L)Testing vs Drug Device Compatibility Studies: Key Differences
| Parameter | E&L Testing | Drug-Device Compatibility Studies |
|---|---|---|
| Primary objective | Identify migrating chemicals | Evaluate overall formulation-device interaction |
| Main concern | Patient chemical exposure | Product performance and stability |
| Focus | Chemical safety | Functional compatibility |
| Evaluates | Extractables, leachables | Drug stability, device performance |
| Regulatory emphasis | Toxicological assessment | Product quality and functionality |
| Typical techniques | LC-MS/MS, GC-MS, ICP-MS | Stability studies, functional testing, mechanical evaluation |
| Risk addressed | Toxic impurities | Device failure or formulation incompatibility |
| Timing | Material qualification and product lifecycle | Development through commercialization |
| Output | Chemical identification and risk assessment | Performance and compatibility report |
When Are Both Studies Needed?
Many product categories require both evaluations simultaneously, including prefilled syringes, autoinjectors, pen injectors, wearable injectors, ophthalmic drug products, nasal sprays, metered-dose and dry powder inhalers, infusion pumps, large-volume parenterals, combination products, and drug-eluting medical devices. Using only one study type leaves a significant regulatory and patient safety gap — chemical safety data alone says nothing about whether a device will deliver an accurate dose after 24 months of storage, and functional data alone says nothing about trace-level chemical exposure.
Regulatory Expectations
Global regulatory authorities increasingly expect comprehensive, risk-based assessments rather than isolated studies. Relevant guidance includes FDA combination product guidance, USP <1663> (Assessment of Extractables), USP <1664> (Assessment of Drug Product Leachables), USP <381> and <382> (Elastomeric Closures and Components), ISO 10993 (Biological Evaluation), and ICH Q8, Q9, and Q10.
The regulatory landscape for E&L is also evolving quickly with the emergence of ICH Q3E, a harmonized guideline specifically addressing extractables and leachables risk assessment. Our detailed breakdowns of ICH Q3E study requirements, ICH Q3E risk assessment methodology, and the ICH Q3E guideline overview walk through how this framework is reshaping E&L expectations globally.
Requirements also differ by market. Sponsors preparing for U.S. approval should review E&L requirements for U.S. market authorization and our broader overview of E&L testing in the United States, while multinational programs benefit from understanding how expectations diverge in our comparison of E&L testing in the USA vs. Europe. For NDA and ANDA-track products specifically, E&L testing for drug safety in NDA/ANDA submissions outlines how E&L data supports the CMC package, and our review of FDA extractables and leachables case studies shows how these expectations have played out in real submissions.
Manufacturers should develop scientifically justified study designs aligned with product risk, dosage form, route of administration, patient population, and storage conditions, rather than defaulting to a generic testing template.
Special Product Categories
Certain product classes carry unique E&L and compatibility considerations that warrant dedicated study design:
- Biologics and advanced therapies: protein and cell-based products are especially sensitive to leachable-driven aggregation and instability. See our guidance on extractables and leachables in biologics and ATMPs, E&L in emerging biologics and advanced therapies, and our broader look at extractables and leachables in biologics.
- Injectable lifecycle products: reformulation or packaging changes for established injectables can reopen E&L questions, as discussed in our case review of extractables and leachables in dexamethasone injectables.
- Veterinary drug products: E&L expectations for veterinary products follow a related but distinct risk framework, covered in E&L testing for veterinary drug products.
- Genotoxic and carcinogenic leachables: certain leachables, including nitrosamines, require specialized toxicological evaluation; see extractables and leachables carcinogenicity testing.
4: Challenges in Extractables and Leachables (E&L) and Drug-Device Compatibility Studies
Both Extractables and Leachables (E&L) testing and drug-device compatibility studies present unique scientific and analytical challenges that can significantly influence product development timelines, regulatory approvals, and patient safety. While E&L studies focus on identifying and assessing chemical migrants from packaging and device materials, compatibility studies evaluate how the drug formulation and delivery device interact over time. Addressing these challenges requires a multidisciplinary approach involving analytical chemistry, materials science, pharmaceutical development, toxicology, and regulatory expertise.
Challenges in E&L testing:
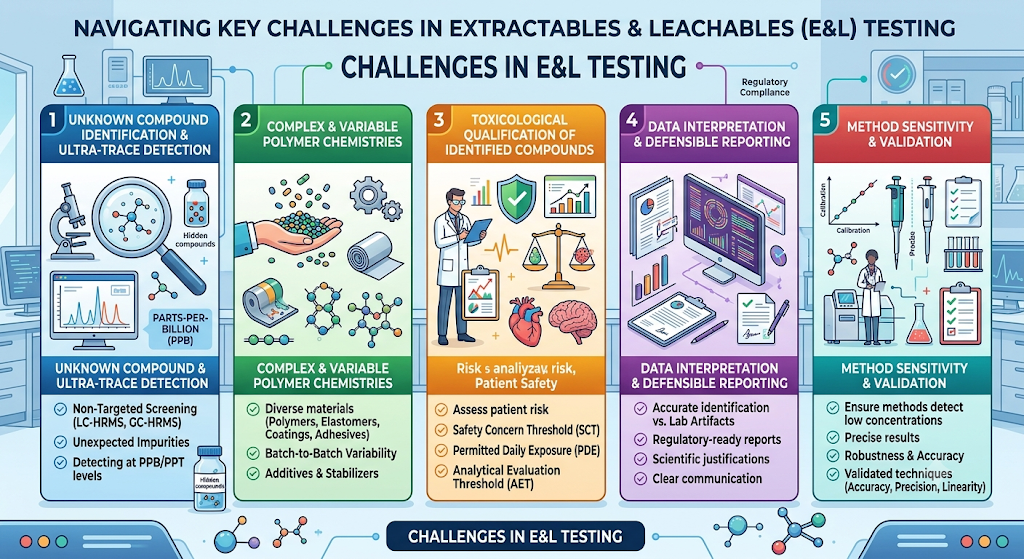
1. Unknown Compound Identification and Ultra-Trace Detection
One of the biggest challenges in E&L testing is identifying unknown compounds present at extremely low concentrations, often in the parts-per-billion (ppb) or parts-per-trillion (ppt) range. Packaging materials can release unexpected impurities that are not included in targeted analytical methods. Advanced techniques such as LC-HRMS and GC-HRMS are often required to perform non-targeted screening and confidently identify these compounds. Detecting and characterizing unknown leachables is critical for accurate toxicological assessment and regulatory compliance.
2. Complex and Variable Polymer Chemistries
Modern pharmaceutical packaging and medical devices are manufactured using a wide range of polymers, elastomers, adhesives, and coatings. Even materials from different suppliers—or different manufacturing lots from the same supplier—can vary in composition due to changes in additives, stabilizers, catalysts, and processing aids. These variations may alter the extractable profile, making study design and data interpretation more complex. Comprehensive material characterization is therefore essential before initiating E&L studies.
3. Toxicological Qualification of Identified Compounds
Identifying a leachable is only the first step. Manufacturers must also determine whether patient exposure to that compound presents an acceptable toxicological risk. This requires estimating daily exposure levels, reviewing available toxicological data, and comparing results against established safety thresholds such as the Analytical Evaluation Threshold (AET), Safety Concern Threshold (SCT), or Permitted Daily Exposure (PDE), where applicable. For novel or previously unreported compounds, additional toxicological assessment may be required.
4. Data Interpretation and Defensible Reporting
Generating analytical data is not enough—results must be scientifically justified and presented in a clear, regulatory-ready format. E&L studies often produce large datasets containing numerous detected compounds, requiring careful interpretation to distinguish true leachables from laboratory artifacts or background contaminants. Accurate compound identification, appropriate risk assessment, and transparent reporting are essential for supporting regulatory submissions and responding to agency questions.
Data interpretation and defensible reporting — a discipline covered in our article on data integrity in extractables and leachables testing
5. Method Sensitivity and Validation
Analytical methods used in E&L testing must be sufficiently sensitive to detect compounds at concentrations relevant to patient safety. Selecting the wrong analytical technique or an inappropriate detection limit can result in missed impurities or unreliable data. Method development and validation should demonstrate accuracy, precision, specificity, linearity, robustness, and appropriate limits of detection and quantification to ensure reliable and reproducible results.
Method sensitivity and validation, addressed in method validation for extractables and leachables testing.
Challenges in Drug-Device Compatibility Studies:
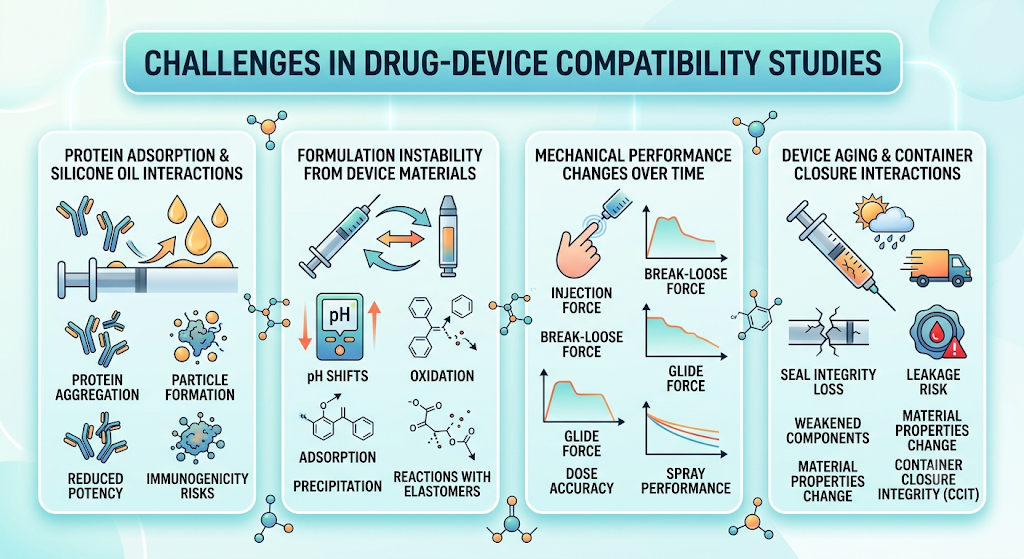
1. Protein Adsorption and Silicone Oil Interactions
Biologics and therapeutic proteins are particularly susceptible to adsorption onto device surfaces or interactions with silicone oil used to lubricate syringes and cartridges. These interactions can lead to protein aggregation, particle formation, reduced potency, or immunogenicity concerns. Compatibility studies must carefully evaluate these effects throughout the product’s shelf life.
2. Formulation Instability Caused by Device Materials
Drug formulations may undergo unexpected chemical or physical changes when stored in prolonged contact with packaging or device materials. Factors such as pH shifts, oxidation, adsorption, precipitation, or reactions with elastomers and plastics can compromise product stability and efficacy. Identifying these interactions early allows manufacturers to optimize material selection and formulation design.
3. Mechanical Performance Changes Over Time
Drug delivery devices must maintain consistent functionality throughout storage and use. Mechanical properties such as injection force, break-loose force, glide force, dose accuracy, spray performance, and residual volume may change as device components age or interact with the formulation. Long-term compatibility studies help ensure reliable device performance until the end of the labeled shelf life.
4. Device Aging and Container Closure Interactions
Environmental conditions such as temperature fluctuations, humidity, transportation stress, and prolonged storage can affect both the delivery device and the container closure system. Aging may alter seal integrity, increase leakage risk, weaken mechanical components, or change material properties. Container closure integrity testing (CCIT) and accelerated aging studies are therefore critical elements of comprehensive compatibility evaluations.
Many of these challenges surface only after a study has already been executed, which is why understanding the root causes of failed E&L studies — inadequate extraction conditions, mismatched analytical sensitivity, or poorly justified thresholds — is as valuable as the study design itself.
5: Best Practices for Successful Study Design
- Perform early risk assessments to identify high-risk materials before formulation development begins.
- Select appropriate, orthogonal analytical methods capable of detecting volatile, semi-volatile, non-volatile, and elemental contaminants.
- Design product-specific studies reflecting the actual drug formulation, route of administration, storage conditions, packaging configuration, and intended shelf life.
- Integrate toxicological assessment so chemical identification is always paired with safety qualification and exposure assessment.
- Conduct lifecycle monitoring across development, validation, commercial manufacturing, and ongoing stability studies.
Why Early Integration Saves Time and Cost
Companies frequently delay compatibility or E&L studies until late-stage development. This approach often leads to unexpected regulatory questions, additional analytical testing, product redesign, packaging changes, stability delays, clinical timeline disruptions, and increased development costs.
Integrating both studies early provides faster regulatory submissions, better risk management, improved patient safety, reduced commercialization delays, and greater confidence in long-term product performance — a shift also reflected in where the field itself is heading, as outlined in our perspective on the future of extractables and leachables testing.
6: How ResolveMass Laboratories Inc. Supports Pharmaceutical Developers
ResolveMass Laboratories Inc. delivers comprehensive analytical services supporting pharmaceutical, biotechnology, and medical device companies throughout product development. Our scientific capabilities include:
- Extractables and leachables study design, including E&L testing services for prefilled syringes
- Unknown compound identification using high-resolution mass spectrometry
- LC-MS/MS, LC-HRMS, GC-MS/MS, GC-HRMS, and ICP-MS analysis
- Toxicological risk assessment support
- Material characterization and stability-indicating analytical methods
- Combination product analytical support
- Regulatory-ready technical reports
- Method development and validation
- Customized, risk-based testing strategies aligned with FDA, USP, EMA, and ICH expectations
For sponsors weighing internal build-out against a specialized partner, our guide on outsourcing E&L testing to a U.S. laboratory outlines what to look for in a contract testing partner’s scientific depth, regulatory experience, and turnaround capability.
By combining advanced analytical technologies with regulatory expertise, ResolveMass helps clients generate high-quality data that supports IND, NDA, ANDA, BLA, and global regulatory submissions.
Conclusion:
Understanding E&L Testing vs Drug Device Compatibility Studies is essential for ensuring the safety, quality, and performance of modern pharmaceutical products and combination devices. Although the two study types are closely related, they address different scientific objectives: E&L testing focuses on identifying and assessing chemical contaminants that migrate from materials, while drug-device compatibility studies evaluate whether the formulation and delivery system function together effectively over the product lifecycle.
For complex products such as biologics, injectable therapies, ophthalmics, inhalation products, and combination devices, integrating both assessments early in development strengthens regulatory compliance, minimizes development risk, and enhances patient safety. Partnering with an experienced analytical laboratory such as ResolveMass Laboratories Inc. ensures scientifically robust, regulatory-ready data that accelerates successful product development.
Frequently Asked Questions:
E&L testing is not mandatory for every pharmaceutical packaging system, but it is strongly recommended or expected for products where there is a significant risk of chemical migration into the drug product. Injectable drugs, biologics, ophthalmic formulations, inhalation products, and combination products typically require comprehensive E&L evaluations. Regulatory authorities expect manufacturers to perform a science-based risk assessment considering the packaging material, route of administration, duration of patient exposure, and storage conditions. High-risk products often require detailed extractables and leachables studies to demonstrate patient safety. The testing strategy should align with USP <1663>, USP <1664>, FDA, EMA, and ICH recommendations. Conducting E&L studies early also helps avoid regulatory questions, product redesigns, and delays during commercialization.
No, E&L testing cannot replace drug-device compatibility studies because both address different aspects of product safety and performance. E&L testing focuses on identifying and quantifying chemicals that migrate from packaging or device materials into the drug product. Drug-device compatibility studies evaluate whether the formulation and delivery device remain physically, chemically, and mechanically compatible over the intended shelf life. They assess parameters such as dose accuracy, drug stability, injection performance, and material interactions. A product may pass E&L testing but still fail compatibility due to device malfunction or formulation instability. Regulatory agencies generally expect both studies for drug-device combination products to provide comprehensive evidence of product quality and patient safety.
Many complex pharmaceutical products require both evaluations to satisfy regulatory expectations and ensure patient safety. These include prefilled syringes, autoinjectors, pen injectors, wearable injectors, ophthalmic solutions, nasal sprays, metered-dose inhalers, dry powder inhalers, infusion systems, biologics, vaccines, and drug-eluting medical devices. These products involve prolonged contact between the formulation and packaging or delivery device, increasing the potential for chemical migration and material interactions. Both studies help confirm that the product remains stable, effective, and safe throughout storage and clinical use. Performing them together provides a complete understanding of the interaction between the formulation and its delivery system.
Several international regulatory guidelines provide recommendations for designing and conducting these studies. USP <1663> addresses extractables assessments, while USP <1664> focuses on leachables evaluations. Additional guidance is available from the FDA for combination products, ISO 10993 for biological evaluation of medical devices, and ICH Q8, Q9, and Q10 for pharmaceutical development and quality risk management. Depending on the dosage form, EMA and other regional agencies may also have product-specific expectations. Manufacturers are encouraged to adopt a risk-based, scientifically justified testing strategy that reflects product characteristics, patient exposure, and intended use. Following these guidelines supports successful regulatory submissions and product approvals.
High-resolution mass spectrometry (HRMS) plays a critical role in modern E&L testing because it enables highly accurate identification of unknown compounds present at extremely low concentrations. Unlike targeted methods, HRMS supports non-targeted screening, allowing scientists to discover unexpected extractables and leachables without prior knowledge of their identity. Accurate mass measurements and isotope pattern analysis improve confidence in structural elucidation. HRMS is particularly valuable for complex polymer systems and combination products where unknown impurities may be present. The technology also supports retrospective data analysis when new regulatory concerns arise. These capabilities make HRMS an indispensable tool for comprehensive E&L investigations.
The ideal time to begin these studies is during the early stages of formulation and packaging development. Initial risk assessments help identify potential concerns before selecting final materials or device components. E&L testing can begin with material qualification, followed by leachables evaluations during stability studies. Drug-device compatibility assessments should continue throughout formulation optimization, engineering studies, process validation, and commercial stability programs. Early integration allows manufacturers to identify issues before regulatory submissions, reducing the likelihood of costly redesigns or additional testing. A proactive strategy also shortens development timelines and improves overall product quality. This lifecycle approach aligns well with current regulatory expectations.
Drug-device compatibility studies assess how well the formulation and delivery system perform together over the product’s intended shelf life. Scientists evaluate drug potency, degradation products, pH, particulate formation, protein aggregation, and overall formulation stability. Device performance testing includes injection force, dose accuracy, spray characteristics, residual volume, mechanical integrity, and container closure performance. Material interactions such as silicone oil compatibility, elastomer swelling, and polymer degradation are also investigated. Stability studies under real-time and accelerated conditions help predict long-term performance. These evaluations ensure that patients receive safe, accurate, and reliable drug delivery throughout the product lifecycle.
Reference
- Xu A, Christiaens P. Extractable and Leachable Challenges in Lyophilized Drug Products. Pharmaceutical Technology. 2025 Apr;49(3):28-30.https://www.pharmtech.com/view/extractable-leachable-challenges-lyophilized-drug-products
- Riter J, Bantz D. Analytical Testing Considerations for Combination Products. The Combination Products Handbook. 2023 May 16:399-418.https://www.taylorfrancis.com/chapters/edit/10.1201/9781003300298-14/analytical-testing-considerations-combination-products-jennifer-riter-daniel-bantz
- Blümel M, Liu J, de Jong I, Weiser S, Fast J, Litowski J, Shuman M, Mehta SB, Amery L, Tan DC, Jia F. Current industry best practice on in-use stability and compatibility studies for biological products. Journal of pharmaceutical sciences. 2023 Sep 1;112(9):2332-46.https://www.sciencedirect.com/science/article/pii/S0022354923001909
- Smith EJ, Paskiet DM, Tullo EJ. The management of extractables and leachables in pharmaceutical products. InParenteral Medications, Fourth Edition 2019 Jul 19 (pp. 535-573). CRC Press.https://www.taylorfrancis.com/chapters/edit/10.1201/9780429201400-29/management-extractables-leachables-pharmaceutical-products-edward-smith-diane-paskiet-erica-tullo

