
Introduction
The regulatory framework governing Extrapolation of Indications in Biosimilar Approval is a well-established, science-driven approach that enables a biosimilar to receive approval for multiple therapeutic indications held by its reference biologic without the need for comparative clinical trials in every individual patient population. This regulatory strategy is founded on the principle that when a biosimilar demonstrates a high degree of similarity to the reference product with respect to its structural composition, biological function, pharmacokinetic behavior, and clinical performance in a single, highly sensitive index indication, it can be expected to deliver equivalent clinical outcomes across all other approved indications of the reference medicine. To satisfy the stringent “totality of evidence” requirements established by regulatory agencies, manufacturers must generate comprehensive analytical datasets using advanced characterization platforms such as high-resolution mass spectrometry, nuclear magnetic resonance (NMR) spectroscopy, and highly sensitive bioassays. For specialized contract research organizations (CROs) supporting biosimilar development, including the USFDA-registered, Health Canada GMP-licensed, and ISO 9001:2015 certified ResolveMass Laboratories Inc., the primary emphasis shifts away from conducting repetitive and expensive clinical studies toward extensive structural, physicochemical, and functional characterization of the biosimilar molecule.
Ensure your development program meets global standards with our comprehensive biosimilar characterization services.
Biological therapeutics represent one of the most sophisticated and expensive categories of medicines within the global healthcare market. Blockbuster biologics such as adalimumab (Humira), which historically exceeded 84,000 USD in annual treatment costs before the introduction of biosimilars, have placed a considerable financial burden on healthcare systems, insurance providers, and patients worldwide. The biosimilar regulatory pathway was specifically developed to foster market competition and improve patient affordability. However, this objective can only be achieved efficiently if manufacturers are able to obtain approval for the complete range of therapeutic indications associated with the reference biologic without repeating large-scale Phase III clinical trials for every individual disease indication. Through scientifically justified extrapolation, sponsors can perform a single confirmatory clinical study in the most sensitive patient population and use the resulting evidence, together with extensive analytical and functional comparability data, to support approval across the entire product label. This streamlined strategy significantly shortens development timelines, lowers research and development expenditures, and ultimately expands patient access to life-changing biological therapies.
Simplify your development path: Learn more about our robust biosimilar comparability studies.
Share via:
Article Summary:
- Extrapolation of indications allows a biosimilar to be approved for multiple therapeutic uses after demonstrating strong similarity to the reference biologic in one sensitive indication, eliminating the need for separate clinical trials for every approved disease.
- The totality of evidence is the cornerstone of biosimilar approval, combining comprehensive analytical characterization, functional testing, pharmacokinetic/pharmacodynamic (PK/PD) studies, immunogenicity assessment, and confirmatory clinical data to establish biosimilarity.
- Advanced analytical techniques such as high-resolution mass spectrometry, NMR spectroscopy, glycan profiling, peptide mapping, and bioassays play a central role in confirming structural, physicochemical, and functional equivalence between the biosimilar and the reference product.
- Mechanism of action and biological function must remain consistent across all proposed indications. Developers demonstrate comparable target binding, receptor interactions, Fc-mediated effector functions, and clinical pharmacology to scientifically justify indication extrapolation.
- Global regulatory agencies, including the EMA, US FDA, and Health Canada, generally support indication extrapolation, although specific evidence requirements may vary depending on regional regulations, product complexity, and therapeutic application.
- Real-world case studies involving biosimilars such as filgrastim, infliximab, rituximab, and trastuzumab illustrate how robust analytical and functional comparability can successfully support approval across multiple diseases without repeating extensive clinical studies.
- As biologics become more complex, including antibody-drug conjugates (ADCs) and bispecific antibodies, developers must generate increasingly sophisticated analytical and functional data. This science-driven approach continues to improve development efficiency, reduce costs, accelerate patient access, and maintain high standards of quality, safety, and efficacy.

Core Scientific Foundations of the Comparative Comparability Exercise
The scientific foundation of the biosimilar comparability exercise is based on the concept that a close structural match results in equivalent biological function and clinical performance. When a biosimilar developer demonstrates that the proposed product possesses an identical primary amino acid sequence and closely matches the structural and functional characteristics of the reference biologic within its naturally occurring lot-to-lot variability, regulators can conclude that its safety, efficacy, and overall clinical performance will remain consistent across different therapeutic indications. This conclusion is supported through a structured, hierarchical evidence package commonly referred to as the “totality of evidence” approach.

Master the requirements for biosimilar comparability studies to streamline your regulatory submission.
Within this evidence hierarchy, analytical characterization carries the greatest scientific weight because it directly establishes molecular similarity between the biosimilar and its reference product. Clinical investigations primarily serve a confirmatory purpose, focusing on identifying any clinically meaningful differences rather than independently proving efficacy or safety.
To accurately define the acceptable variability of the reference biologic, sponsors undertake a comprehensive reverse-engineering strategy. Numerous batches of the originator product are collected from major international markets, including the United States, Europe, and Japan, over an extended period. Detailed evaluation of these batches enables developers to establish the acceptable ranges for Critical Quality Attributes (CQAs), allowing manufacturing processes to be optimized so that the biosimilar consistently falls within the natural variability demonstrated by the reference product.
Identify and monitor critical quality attributes (CQAs) in biosimilars to ensure consistent product quality.
Analytical Validation in the Extrapolation of Indications in Biosimilar Approval
Analytical validation for Extrapolation of Indications in Biosimilar Approval relies on a comprehensive collection of orthogonal analytical techniques designed to confirm that the biosimilar exhibits physicochemical and structural characteristics equivalent to those of the reference biologic. Since therapeutic monoclonal antibodies are highly complex glycoproteins with an approximate molecular weight of 148 kDa, developers must demonstrate that minor microheterogeneity does not influence biological activity, safety, or clinical performance. Consequently, multiple complementary analytical platforms are employed to evaluate the molecule from several structural and functional perspectives.
| Characterization Parameter | Target of Analysis | Primary Analytical Technologies | Clinical & Functional Relevance |
|---|---|---|---|
| Primary Structure | Amino acid sequence and sequence variants | High-resolution LC-MS/MS peptide mapping and sequence confirmation | Confirms an identical protein backbone while identifying sequence modifications such as deamidation and oxidation. |
| Secondary & Tertiary Structure | Protein folding, higher-order structure, and solvent accessibility | Circular Dichroism (CD), FTIR, Differential Scanning Calorimetry (DSC), and HDX-MS | Demonstrates appropriate three-dimensional conformation and verifies thermal stability. |
| Glycan Profiling | N-glycosylation patterns at the conserved Asn-297 residue | HILIC-FLD, mass spectrometric glycan mapping, and Capillary Electrophoresis | Influences Fcγ receptor interactions and determines ADCC and CDC functional activity. |
| Purity & Heterogeneity | Protein aggregates, charge variants, and molecular fragments | SEC-MALS and capillary isoelectric focusing (cIEF/icIEF) | Minimizes the risk of immunogenicity associated with protein aggregation and structural heterogeneity. |
| Process Impurities | Host cell proteins (HCPs) and residual host cell DNA | ELISA, quantitative PCR (qPCR), and process-specific microfluidic analytical methods | Confirms product purity while reducing the potential for process-related immunogenic responses. |
Utilize advanced proteomics approach for biosimilars to ensure deep molecular characterization.
The sensitivity and resolution of modern analytical technologies have advanced substantially, enabling developers to define structure-function relationships with an exceptional level of precision. These technological improvements provide regulatory authorities with highly reliable analytical evidence supporting biosimilar comparability, thereby allowing indication extrapolation to be justified primarily through robust laboratory characterization rather than relying extensively on comparatively less sensitive clinical trials.
Leverage native mass spectrometry for biosimilars to analyze higher-order structures with superior accuracy.
Mechanisms of Action and Reconstitution of Cellular Functionality
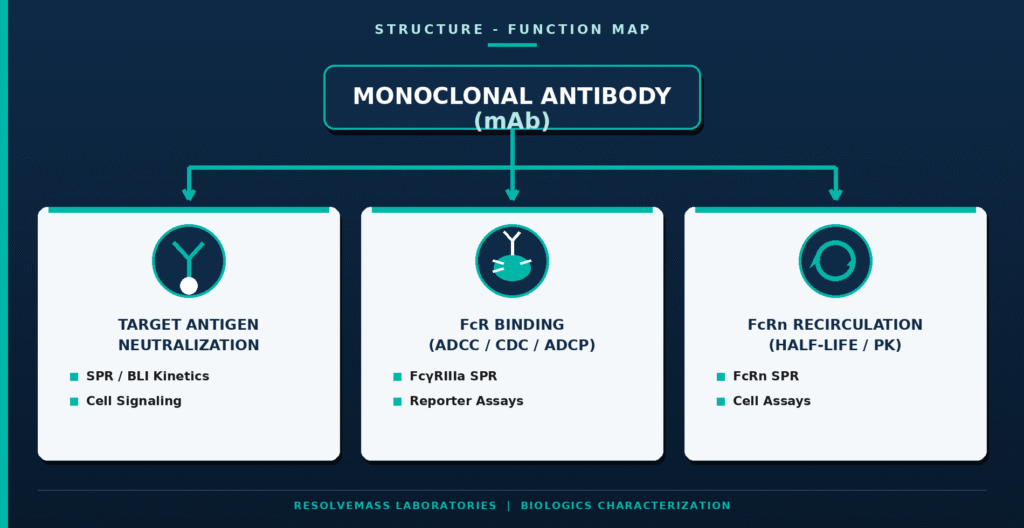
The scientific basis for Extrapolation of Indications in Biosimilar Approval depends on demonstrating that the primary mechanisms of action (MoAs) and the relevant target receptors remain consistent between the clinically studied indication and every indication proposed for extrapolation. For complex biologics that exhibit multiple mechanisms of action across different disease settings, sponsors are required to establish functional equivalence for each individual biological pathway before extrapolation can be scientifically justified.
Ensure your product meets safety standards with our impurity profiling of biosimilars expertise.
To comprehensively evaluate the molecule’s functional characteristics, developers employ several specialized analytical and cell-based assays capable of measuring different biological activities.
Target Binding and Neutralization: Surface Plasmon Resonance (SPR) and Biolayer Interferometry (BLI) are used to determine binding affinity (Kd) together with the kinetic constants (kon and koff). Complementary cell-based bioassays assess the molecule’s capacity to inhibit downstream inflammatory or proliferative signaling pathways, thereby confirming functional equivalence with the reference biologic.
Fc-Mediated Effector Functions: Antibody-Dependent Cellular Cytotoxicity (ADCC) is assessed using either primary natural killer (NK) cells or genetically engineered reporter cell systems. Complement-Dependent Cytotoxicity (CDC) assays measure complement activation, while Antibody-Dependent Cellular Phagocytosis (ADCP) assays evaluate macrophage-mediated clearance. Collectively, these studies verify that Fc-mediated biological functions are comparable between the biosimilar and the reference product.
Neonatal Fc Receptor (FcRn) Binding: SPR-based FcRn binding studies investigate the pH-dependent interaction between the antibody and the neonatal Fc receptor. Because FcRn recycling plays a central role in determining antibody half-life and systemic exposure, these assays provide valuable insight into expected pharmacokinetic performance in human patients.
Investigate the impact of molecular changes using charge variant analysis in biosimilars
and aggregation analysis in biosimilars.
When the target antigen is expressed differently across tissues or when disease pathophysiology varies between therapeutic indications, the biosimilar comparability exercise must demonstrate that the molecule retains identical biological activity under all relevant physiological conditions. Establishing this functional consistency provides a critical scientific basis for extrapolating clinical efficacy and safety beyond the directly studied indication.
Understand regulatory expectations by reviewing ICH Q6B guidelines for biological characterisation.
Comparative Clinical Pharmacology and Next-Generation Immunogenicity Mapping
Comparative clinical pharmacology studies serve as the essential bridge between comprehensive laboratory characterization and clinical performance by confirming pharmacokinetic (PK) and pharmacodynamic (PD) equivalence in carefully selected, highly sensitive study populations. Pharmacokinetic comparability is generally established through randomized, double-blind Phase I clinical studies, where the 90% confidence intervals for Cmax, AUCt, and AUC0–∞ are required to remain within the accepted bioequivalence range of 80–125%.
Beyond traditional clinical pharmacology studies, biosimilar developers increasingly incorporate next-generation immunogenicity assessment strategies. Since immunogenicity is strongly influenced by patient-specific immune responses, disease characteristics, and treatment regimens, thorough evaluation of in vivo immune responses remains a critical component of biosimilar development.
Modern computational platforms integrate machine learning algorithms with advanced structural prediction tools—including EpiMatrix, JanusMatrix, and the Immune Epitope Database (IEDB)—to identify peptide sequences that are likely to bind HLA Class II molecules and stimulate CD4+ T-helper cell responses. When combined with comprehensive clinical monitoring of anti-drug antibodies (ADAs), these predictive epitope-mapping approaches provide a robust immunogenicity assessment capable of supporting scientifically justified extrapolation from lower-risk populations to patient groups with greater immunological complexity.
Regulatory Precedents and Regional Divergence in Approvals
Although the fundamental scientific principles supporting biosimilar comparability are widely accepted worldwide, individual regulatory agencies implement these principles within distinct legal, public health, and clinical frameworks. Consequently, regional differences in approved indications may arise despite similar scientific evidence. These variations are commonly influenced by national legislation, intellectual property considerations, regulatory philosophy, and differing approaches to managing scientific uncertainty.
| Regulatory Authority | Key Statutory Pathways | Core Approach to Extrapolation | Notable Exceptions & Nuances |
|---|---|---|---|
| EMA (Europe) | Article 10(4) of Directive 2001/83/EC | Strongly supports extrapolation when comprehensive structural and functional similarity has been demonstrated. | Established the first biosimilar approval pathway and possesses more than two decades of regulatory and clinical experience. |
| US FDA (United States) | Section 351(k) of the Public Health Service (PHS) Act under the BPCIA | Reviews extrapolation requests individually based on the strength of the total scientific evidence. | Introduces an additional “interchangeability” designation that permits pharmacy-level substitution under specific regulatory requirements. |
| Health Canada (Canada) | New Drug Submission (NDS) pathway | Supports extrapolation while applying additional scrutiny to products with particularly complex mechanisms of action. | Historically withheld inflammatory bowel disease (IBD) indications for biosimilar infliximab until additional bridging evidence became available. |
When planning global biosimilar development programs, sponsors must carefully consider these regional regulatory differences. For instance, while the European Medicines Agency (EMA) may approve all therapeutic indications based primarily on extensive analytical and functional comparability data, the US FDA or Health Canada may request additional pharmacokinetic, pharmacodynamic, or clinical bridging studies for selected target populations before granting full indication extrapolation.
Drug-Specific Case Studies in Indication Extrapolation
Filgrastim: The Template for Extrapolation of Indications in Biosimilar Approval
Filgrastim became one of the earliest biologics to receive worldwide biosimilar approval across all licensed indications through the indication extrapolation framework. As a nonglycosylated recombinant human granulocyte colony-stimulating factor (G-CSF), filgrastim promotes neutrophil recovery following chemotherapy and stimulates the mobilization of peripheral blood progenitor cells in both patients and healthy donors. Owing to its relatively simple protein structure and the absence of glycosylation, filgrastim can be characterized with a very high degree of analytical confidence.
To justify extrapolation for healthy donor stem-cell mobilization, developers demonstrated that the biosimilar possessed binding characteristics equivalent to those of the reference product at the G-CSF receptor. This equivalent receptor interaction produced identical downstream intracellular signaling and comparable biological mobilization of hematopoietic progenitor cells regardless of the target population. Based on this comprehensive structural and functional comparability, regulatory authorities concluded that additional indication-specific clinical trials were unnecessary and granted approval across all approved therapeutic indications of the reference product.
Explore specialized analytical workflows for insulin biosimilar characterization.
Infliximab: Overcoming the Antibody-Dependent Cellular Cytotoxicity Divide
The regulatory evaluation of the infliximab biosimilar CT-P13 represents one of the most influential examples of indication extrapolation, particularly in extending approval from rheumatologic diseases to inflammatory bowel disease (IBD). Infliximab is a chimeric IgG1 monoclonal antibody directed against both soluble and membrane-bound tumor necrosis factor-alpha (TNF-α). During analytical characterization, investigators identified a minor 2% reduction in afucosylated glycan species in CT-P13 compared with the reference product (Remicade). This subtle glycosylation difference resulted in slightly reduced in vitro binding affinity to the FcγRIIIa receptor and correspondingly lower ADCC activity under simplified laboratory assay conditions.
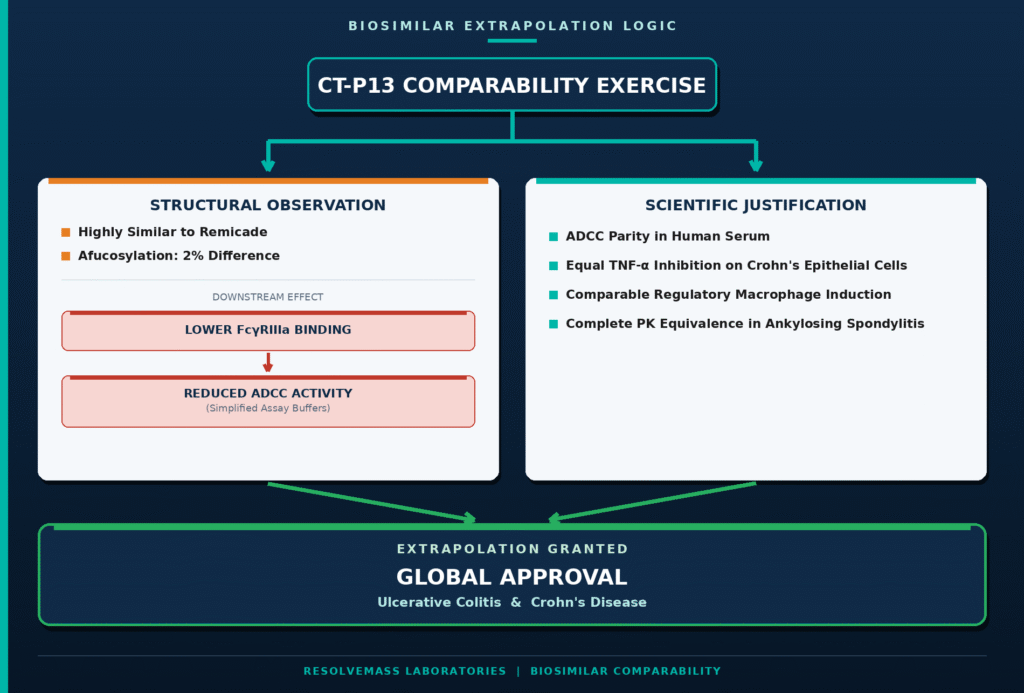
To determine whether this analytical difference had any clinical significance, the biosimilar developer performed additional physiological and functional investigations. These studies demonstrated several important findings:
- The apparent reduction in ADCC activity disappeared when assays were conducted under physiologically relevant conditions using human serum.
- Both the biosimilar and the reference product exhibited equivalent inhibition of TNF-α signaling in epithelial cell models and produced comparable induction of regulatory macrophages, two mechanisms considered central to therapeutic efficacy in Crohn’s disease and ulcerative colitis.
- Comparative multiple-dose pharmacokinetic studies involving 250 patients with active ankylosing spondylitis confirmed equivalent systemic exposure, indicating that patients with inflammatory bowel disease would be expected to experience similar pharmacokinetic profiles.
Considering this comprehensive totality of evidence, both the EMA and the US FDA approved extrapolation of CT-P13 for all inflammatory bowel disease indications. Although Health Canada initially deferred approval for IBD because of concerns regarding the contribution of ADCC to intestinal mucosal healing, the agency granted full authorization in 2016 after reviewing additional analytical, functional, and clinical bridging data submitted by the sponsor.
Stay ahead of potential stability issues with our forced degradation of biosimilars studies.
Rituximab: Bridging Autoimmune and Oncological Environments
The extrapolation of biosimilar rituximab products, including SDZ-RTX and Truxima, from rheumatoid arthritis (RA) to non-Hodgkin lymphoma (NHL) illustrates the scientific complexity involved in extending approval across substantially different therapeutic settings. Rituximab is a chimeric IgG1 monoclonal antibody that specifically targets the CD20 antigen expressed on B lymphocytes, resulting in B-cell depletion through multiple complementary mechanisms, including Antibody-Dependent Cellular Cytotoxicity (ADCC), Complement-Dependent Cytotoxicity (CDC), Antibody-Dependent Cellular Phagocytosis (ADCP), and direct induction of apoptosis.
During regulatory evaluation, the biosimilar established clinical efficacy and safety in a pivotal study involving patients with active rheumatoid arthritis. However, extending approval to oncology indications required developers to scientifically address several important pharmacological and clinical differences between autoimmune disorders and hematological malignancies.
Target Tissue Characteristics: In rheumatoid arthritis, rituximab primarily acts on normal circulating B lymphocytes involved in autoimmune inflammation. In contrast, patients with non-Hodgkin lymphoma present with malignant B-cell populations that proliferate rapidly and frequently demonstrate variable levels of CD20 expression. Therefore, developers needed to confirm that equivalent CD20 binding and biological activity were maintained regardless of the cellular environment.
Systemic Pharmacokinetics: Rituximab exhibits distinct pharmacokinetic behavior across autoimmune and oncological diseases. In lymphoma, the presence of a substantial tumor burden creates an antigen sink effect, resulting in accelerated initial drug clearance and greater variability in serum concentrations. Conversely, patients with autoimmune diseases generally display more stable pharmacokinetic profiles because the circulating B-cell population is considerably smaller. These differences required comprehensive pharmacokinetic evaluation to demonstrate comparable drug exposure across diverse patient populations.
Immunogenicity Risk: The immune status of autoimmune and oncology patients differs considerably, resulting in variations in anti-drug antibody (ADA) development. Clinical studies have reported ADA formation in approximately 15–17% of immunocompetent patients with rheumatoid arthritis, whereas immunocompromised patients with lymphoma typically exhibit ADA rates below 1%. Consequently, sponsors were required to provide extensive immunogenicity assessments to demonstrate that these differences would not influence biosimilar performance across extrapolated indications.
To bridge these biological and clinical differences, developers generated detailed receptor-binding studies together with cell-based potency assays demonstrating identical CD20 target engagement and equivalent effector functions in both healthy and malignant B-cell models. Additional comparative clinical investigations in patients with follicular lymphoma confirmed no clinically meaningful differences in pharmacokinetics, safety, efficacy, or overall response rates. Collectively, this comprehensive body of evidence enabled regulatory authorities to approve rituximab biosimilars for both autoimmune and oncological indications through scientifically justified extrapolation.
Ensure your manufacturing process is robust from the start with expert cell line development for biosimilars.
Trastuzumab: Breast Cancer to Gastric Cancer Extrapolation
Biosimilar trastuzumab products, including CT-P6, Ogivri, and SB3, received approval for metastatic gastric cancer entirely through the extrapolation pathway, using comparative clinical evidence generated in patients with HER2-positive early breast cancer. This regulatory decision was supported by the fact that trastuzumab exerts its therapeutic effect through the same fundamental biological mechanism across both disease settings—binding to the extracellular domain of the HER2 receptor and inhibiting downstream cellular signaling pathways. Since this mechanism remains consistent regardless of tissue origin, the scientific rationale for indication extrapolation was considered robust.
Comparative clinical studies for trastuzumab biosimilars are generally conducted in the neoadjuvant breast cancer setting because this model provides a highly sensitive clinical environment for detecting any potential differences between the biosimilar and the reference product. The primary endpoint commonly employed is pathological complete response (pCR), a validated short-term binary endpoint capable of identifying even subtle differences in therapeutic performance. In one pivotal comparative study evaluating the trastuzumab biosimilar SB3 (Ontruzant), the pCR rate reached 51.7% for the biosimilar compared with 42.0% for the reference trastuzumab, with the observed difference remaining well within predefined equivalence margins. These findings provided strong clinical support for extending approval to all other licensed oncological indications.
Subsequent real-world evidence and post-marketing surveillance further strengthened the scientific validity of this extrapolation strategy. In a prospective Japanese post-marketing surveillance study involving 171 patients with HER2-positive advanced gastric cancer treated with CT-P6, investigators observed a disease control rate of 82.4% together with a median progression-free survival (PFS) of 7.4 months. Importantly, no new safety signals or unexpected immunogenicity concerns were identified during treatment. These real-world clinical outcomes closely mirrored those reported for the reference product (Herceptin) in the landmark ToGA clinical trial, providing additional confirmation that extrapolation from breast cancer to gastric cancer was scientifically justified.
Extrapolation Challenges in Next-Generation Therapeutic Modalities
The emergence of next-generation biologics, including antibody-drug conjugates (ADCs) and bispecific antibodies, introduces new scientific challenges to the established principles of indication extrapolation. Unlike conventional monoclonal antibodies, these highly engineered therapeutic molecules incorporate multiple functional domains or chemically linked payloads whose biological behavior may vary depending on the target tissue, cellular environment, or disease indication. Consequently, demonstrating biosimilar comparability for these advanced therapeutic platforms requires considerably more extensive analytical and functional characterization.
| Therapeutic Class | Molecular Structure | Key Extrapolation Challenges | Primary Analytical Requirements |
|---|---|---|---|
| Monoclonal Antibodies | Homogeneous glycoproteins (~148 kDa) | Maintaining glycan-dependent effector functions, including ADCC and CDC, across different tissues | Comprehensive primary structure and higher-order structure (HOS) characterization together with target-binding assays |
| Antibody-Drug Conjugates | Monoclonal antibodies chemically linked to cytotoxic payloads | Demonstrating equivalent conjugation sites, linker stability, and drug-to-antibody ratio (DAR) | Quantification of free and conjugated payloads, evaluation of linker stability under physiological conditions, and cell-based cytotoxicity assays |
| Bispecific Antibodies | Engineered antibodies containing dual-target binding domains | Simultaneous target engagement, structural alignment, and variable inter-target spatial interactions | Measurement of dual binding affinities, simultaneous epitope-binding studies, and target-mediated cellular bridging assays |
For antibody-drug conjugates such as trastuzumab emtansine and trastuzumab deruxtecan, the biosimilar comparability assessment extends far beyond conventional antibody characterization. Sponsors must demonstrate equivalence across several additional synthetic and biological attributes.
Drug-to-Antibody Ratio (DAR): The distribution of cytotoxic payload molecules attached to each antibody molecule must closely match that of the reference product, since deviations in DAR can significantly influence systemic pharmacokinetics, therapeutic efficacy, and toxicity.
Linker Stability: The chemical linker connecting the antibody to its cytotoxic payload must exhibit comparable stability under physiological conditions. Equivalent linker performance is essential to prevent premature release of the cytotoxic drug into systemic circulation, thereby preserving both efficacy and safety.
Payload Potency: The cytotoxic small-molecule payload itself must demonstrate biological activity equivalent to that of the reference product through detailed potency and cell-based cytotoxicity studies.
Similarly, bispecific antibodies such as emicizumab and blinatumomab require developers to establish equivalent simultaneous binding to both molecular targets while demonstrating comparable downstream biological signaling mediated by each binding domain. Because these sophisticated biologics are particularly sensitive to even subtle structural variations, sponsors depend on advanced analytical characterization, high-resolution structural analysis, and comprehensive functional assays to minimize residual uncertainty and provide robust scientific support for their extrapolation submissions.
Conclusion
In summary, the regulatory framework governing Extrapolation of Indications in Biosimilar Approval represents a highly established, science-based approach that enables the efficient development and commercialization of affordable biological therapies. By emphasizing comprehensive analytical characterization, functional comparability, and mechanistic understanding instead of repetitive clinical studies, this pathway significantly accelerates biosimilar development while maintaining rigorous standards for quality, safety, and efficacy. As analytical technologies continue to evolve and achieve increasingly higher levels of sensitivity and precision, the scientific foundation supporting indication extrapolation will rely even more heavily on sophisticated laboratory-based structural and functional characterization.
Successfully navigating this evolving regulatory environment requires specialized scientific expertise and advanced analytical capabilities. Contract research organizations play an essential role in generating the comprehensive comparability data required for global regulatory submissions. ResolveMass Laboratories Inc. supports biosimilar development programs through ISO 9001:2015 certified quality systems, USFDA-registered analytical facilities, and Health Canada GMP-licensed laboratories. By offering advanced services in high-resolution mass spectrometry, custom synthesis, and NMR-based molecular characterization, the laboratory enables developers to generate scientifically robust, structurally comprehensive, and functionally validated comparability packages that support successful biosimilar approval across multiple therapeutic indications.
For inquiries regarding high-resolution mass spectrometry, polymer synthesis, peptide characterization, and analytical support for complex drug development pipelines, contact the scientific team at ResolveMass Laboratories Inc. directly at: https://resolvemass.ca/contact/
Frequently Asked Questions
The most sensitive indication is selected based on its ability to effectively detect potential differences between the biosimilar and reference product in terms of efficacy, safety, and immunogenicity. Selection criteria typically include a well-defined patient population, minimal variability in disease characteristics, and highly responsive clinical endpoints. For example, pathological complete response (pCR) in neoadjuvant oncology studies is considered a sensitive endpoint because it can identify subtle differences in therapeutic activity.
Immunogenicity evaluation is a crucial component of biosimilar development because biologic medicines can stimulate immune responses leading to the formation of anti-drug antibodies (ADAs). These antibodies may reduce therapeutic effectiveness, alter pharmacokinetics, or contribute to safety concerns. Since immune responses can vary depending on disease condition, patient characteristics, and treatment duration, developers must provide strong scientific evidence demonstrating comparable immunogenicity profiles across all extrapolated indications.
Yes, biosimilars approved through extrapolation demonstrate comparable safety, efficacy, and clinical performance to their reference biologics. Regulatory approval is based on an extensive head-to-head comparability program involving advanced analytical characterization, functional testing, pharmacokinetic evaluation, and clinical studies. Extrapolation is only granted when regulatory agencies determine that no meaningful differences exist between the biosimilar and reference product.
Post-translational modifications, including N-glycosylation, play an essential role in determining a biologic’s three-dimensional structure, stability, half-life, and biological activity. Variations in glycan patterns can influence Fc receptor interactions, immune effector functions, and overall therapeutic performance. Therefore, advanced glycan profiling and high-resolution analytical techniques are required to confirm that these modifications remain comparable between the biosimilar and reference product.
Health Canada approval of a biosimilar confirms that the product is highly similar to the reference biologic and has no clinically meaningful differences in quality, safety, or efficacy. However, approval through extrapolation does not automatically establish interchangeability or pharmacy-level substitution. Decisions regarding substitution policies are managed independently by individual Canadian provinces and territories based on their specific healthcare regulations.
Critical Quality Attributes (CQAs) are measurable physical, chemical, biological, or microbiological characteristics that must remain within predefined acceptance limits to ensure the quality, safety, and potency of a biologic product. These attributes include primary amino acid sequence, higher-order structure, glycosylation patterns, purity profile, aggregation levels, and biological activity. Maintaining comparable CQAs between the biosimilar and reference product is essential for demonstrating molecular similarity.
Advanced analytical characterization techniques provide a detailed molecular profile of the biosimilar and reference biologic, enabling precise comparison of their structural and functional properties. Technologies such as LC-MS/MS, Circular Dichroism (CD), Surface Plasmon Resonance (SPR), and capillary isoelectric focusing (icIEF) help identify potential differences in protein structure, binding activity, and product heterogeneity. These analytical datasets form the foundation for scientifically justified indication extrapolation.
Initial concerns regarding infliximab biosimilar extrapolation into inflammatory bowel disease (IBD) were related to observed differences in glycosylation patterns, particularly afucosylation levels, and their potential impact on FcγRIIIa binding and Antibody-Dependent Cellular Cytotoxicity (ADCC). Regulatory authorities questioned whether these variations could influence therapeutic mechanisms involved in Crohn’s disease and ulcerative colitis. Additional functional, physiological, and clinical bridging studies were required to confirm that these differences did not affect clinical outcomes.
Real-world evidence (RWE) provides additional confirmation of the long-term safety, effectiveness, and tolerability of biosimilars after regulatory approval. Post-marketing surveillance studies and observational clinical data help evaluate biosimilar performance in diverse patient populations under routine healthcare conditions. These findings strengthen confidence among healthcare professionals and support the continued use of extrapolated biosimilar indications.
Reference:
- Tesser, J. R. P., Furst, D. E., & Jacobs, I. (2017). Biosimilars and the extrapolation of indications for inflammatory conditions. Biologics: Targets and Therapy, 11, 5–11. https://doi.org/10.2147/BTT.S124476
- Vulto, A. G. (2019). Biologicals and biosimilars in hematology: The case of rituximab. HemaSphere, 3(6), e322. https://doi.org/10.1097/HS9.0000000000000322
- European Medicines Agency. (2012). Guideline on similar biological medicinal products containing monoclonal antibodies—Non-clinical and clinical issues (EMA/CHMP/BMWP/403543/2010). Committee for Medicinal Products for Human Use (CHMP). European Medicines Agency. https://www.ema.europa.eu/en/documents/scientific-guideline/guideline-similar-biological-medicinal-products-containing-monoclonal-antibodies-non-clinical-and-clinical-issues_en.pdf
- Ehmann, F. (2014). EU biosimilar regulatory framework: Non-clinical and clinical aspects. European Medicines Agency. https://www.ema.europa.eu/en/documents/presentation/presentation-eu-biosimilar-regulatory-framework-non-clinical-and-clinical-aspects-iii-falk-ehmann-ema_en.pdf
- Lee, S., Lee, H., & Kim, E. (2019). Comparative efficacy and safety of biosimilar rituximab and originator rituximab in rheumatoid arthritis and non-Hodgkin’s lymphoma: A systematic review and meta-analysis. BioDrugs, 33(5), 469–483. https://doi.org/10.1007/s40259-019-00376-z
- Zhou, X., Yu, J., Wang, W., Song, G., Wang, X., Ren, J., Di, L., & Wang, X. (2015). A phase I dose-escalation study of a biosimilar trastuzumab in Chinese metastatic breast cancer patients. SpringerPlus, 4, Article 803. https://doi.org/10.1186/s40064-015-1603-5

