
Introduction:
When comparing ICH M7 vs FDA Nitrosamine Guidance, both share the same objective — protecting patients from potentially carcinogenic impurities — but they approach the problem differently. ICH M7 provides the overarching scientific framework for assessing and controlling mutagenic impurities throughout drug development, while FDA Nitrosamine Guidance expands that framework with detailed recommendations specific to nitrosamines, reflecting lessons learned from global drug recalls involving sartans, metformin, ranitidine, rifampicin, varenicline, and other pharmaceutical products.
For pharmaceutical manufacturers, understanding both frameworks is essential to building nitrosamine control strategies that satisfy regulators across multiple regions at once, rather than reworking a strategy separately for each market.
Summary:
- ICH M7 vs FDA Nitrosamine Guidance is one of the most common regulatory comparisons pharmaceutical companies face when developing APIs and drug products for global markets.
- ICH M7 establishes a broad, harmonized, risk-based framework for controlling all mutagenic impurities, while FDA nitrosamine guidance narrows the focus to product-specific recommendations for nitrosamine risk assessment, testing, and mitigation.
- Both frameworks share the same underlying goal — minimizing patient exposure to carcinogenic impurities — but differ in scope, implementation detail, and regulatory expectations.
- FDA guidance expands on ICH M7 with detailed acceptable intake (AI) values, confirmatory testing expectations, root cause investigation requirements, and lifecycle management obligations.
- Understanding how the two frameworks align helps sponsors build CMC documentation that holds up across NDA, ANDA, BLA, and international submissions.
- Working with an experienced analytical CRO such as ResolveMass Laboratories Inc. helps pharmaceutical companies generate scientifically sound, regulatory-compliant nitrosamine risk assessments and testing data.
1: Why Nitrosamine Impurities Became a Global Regulatory Priority
Nitrosamine impurities became a global regulatory priority because several widely used medicines were found to contain unacceptable levels of these highly potent mutagenic compounds, triggering recalls and a wave of new guidance. A detailed nitrosamine drug recall analysis shows how findings in sartans, ranitidine, metformin, and rifampin-class products reshaped global expectations for impurity control almost overnight.
Many nitrosamines are classified as probable human carcinogens because long-term, low-level exposure may increase cancer risk. Regulators including FDA, EMA, Health Canada, MHRA, and PMDA now require pharmaceutical companies to proactively identify, assess, test, control, and continuously monitor nitrosamine risks throughout the entire product lifecycle — not just at initial submission.
2: What Is ICH M7?
ICH M7 provides a harmonized international guideline for assessing and controlling DNA-reactive (mutagenic) impurities to limit potential carcinogenic risk in pharmaceuticals. Published by the International Council for Harmonisation (ICH), M7 applies throughout pharmaceutical development and commercial manufacturing, and its most recent update, ICH M7(R2), added a compound-specific acceptable intake addendum that continues to shape how sponsors approach nitrosamine risk. The impact of ICH M7(R2) updates on nitrosamine risk assessment has been significant, particularly for sponsors who built earlier risk assessments around the original 2014 framework.
ICH M7’s primary objectives include:
- Identification of mutagenic impurities
- Structure-based hazard assessment
- Risk categorization (Class 1 through 5)
- Acceptable intake calculations
- Control strategy development
- Lifecycle impurity management
Rather than focusing only on nitrosamines, ICH M7 covers all DNA-reactive mutagenic impurities, including those arising from nitrosamine formation pathways during API synthesis, degradation, starting materials, and reagents.
3: What Is FDA Nitrosamine Guidance?
FDA Nitrosamine Guidance specifically addresses nitrosamine impurities in APIs and finished drug products by expanding risk assessment, testing, reporting, and mitigation expectations well beyond the general ICH M7 framework. The guidance was developed after widespread nitrosamine-related recalls and provides practical, product-level recommendations for manufacturers.
FDA guidance focuses on:
- Nitrosamine risk assessment
- Confirmatory analytical testing
- Root cause investigation
- Acceptable intake limits
- Control strategies
- Supplier qualification
- Process optimization
- Packaging assessment
- Ongoing lifecycle monitoring
Unlike ICH M7, FDA guidance is centered specifically on nitrosamine and NDSRI (nitrosamine drug substance-related impurity) control — understanding the difference between NDSRIs and simple nitrosamines is often the first step in scoping an FDA-aligned risk assessment correctly.
4: ICH M7 vs FDA Nitrosamine Guidance: Quick Comparison
| Parameter | ICH M7 | FDA Nitrosamine Guidance |
|---|---|---|
| Primary Focus | DNA-reactive mutagenic impurities | Nitrosamine impurities |
| Scope | Broad | Nitrosamine-specific |
| Risk Assessment | Required | Required with additional product-level detail |
| Analytical Testing | Risk-based | Strong emphasis on confirmatory testing |
| Acceptable Intake | TTC and compound-specific | AI values published for individual nitrosamines |
| Root Cause Investigation | General | Highly detailed |
| Lifecycle Monitoring | Included | Strong ongoing expectation |
| Packaging Assessment | Limited | Specifically emphasized |
| Supplier Evaluation | General GMP expectations | Detailed evaluation expected |
| Regulatory Submission | Global harmonized framework | FDA-specific implementation |
5: Where Do the Two Frameworks Align?
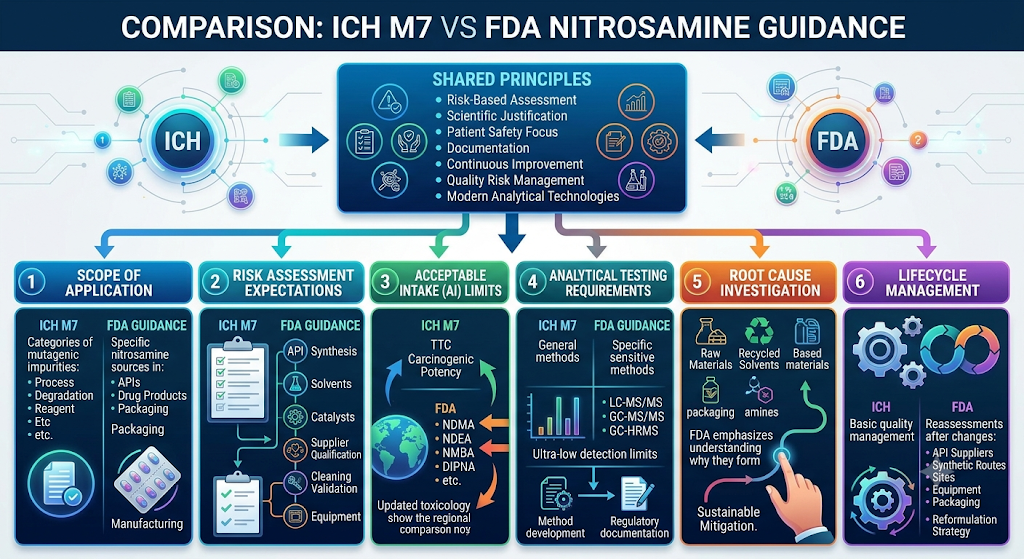
Although different in scope, ICH M7 vs FDA Nitrosamine Guidance reveals substantial regulatory alignment. Both frameworks require pharmaceutical companies to implement science-based impurity control programs built on shared principles: risk-based assessment, scientific justification, impurity identification, patient safety focus, control strategy development, documentation, lifecycle management, quality risk management, and continuous improvement.
Both frameworks also encourage manufacturers to use modern analytical technologies capable of detecting impurities at extremely low concentrations, and both recognize that a nitrosamine impurity arising in an API is not necessarily governed by the same expectations as a nitrosamine leachable originating from packaging — a distinction that matters for how each is investigated and controlled.
Key Difference 1: Scope of Application
The biggest distinction between the two frameworks is scope. ICH M7 applies broadly to mutagenic process impurities, degradation impurities, starting material impurities, reagent-derived impurities, and intermediate impurities — nitrosamines represent only one category of concern within this larger framework.
FDA guidance, by contrast, applies specifically to nitrosamines arising from APIs, drug products, manufacturing processes, and packaging interactions, including nitrosamines formed from excipients, storage-related formation, and secondary nitrosamine formation over shelf life. Packaging-related risk is a particular area of FDA emphasis, covering both packaging leachables and nitrosamine E&L studies broadly and more specific concerns such as nitrosamine leachables in blister packaging. This gives FDA guidance much deeper nitrosamine-specific recommendations than the general ICH M7 text.
Key Difference 2: Risk Assessment Expectations
Both documents require risk assessments, but FDA expects considerably more product-specific detail. An FDA nitrosamine risk assessment typically evaluates API synthesis routes, starting materials, solvents, catalysts, reagents, water systems, packaging materials, storage conditions, manufacturing equipment, cleaning validation, cross-contamination pathways, and contract manufacturers.
A significant part of this work involves solvent and catalyst mitigation strategies to reduce the likelihood of nitrosamine formation at the source, along with structured nitrosamine control during supplier qualification to ensure raw material and intermediate suppliers aren’t introducing undisclosed risk. The goal across all of these activities is identifying every plausible nitrosamine formation pathway before it becomes a confirmed finding.
Key Difference 3: Acceptable Intake (AI) Limits
ICH M7 introduces foundational concepts such as the Threshold of Toxicological Concern (TTC), compound-specific acceptable intake, and carcinogenic potency classification. FDA guidance builds directly on this by publishing AI limits for numerous individual nitrosamines — including NDMA, NDEA, NMBA, DIPNA, EIPNA, NMPA, and NDBA — based on updated toxicological evaluations that FDA revises as new data becomes available.
Applying these limits correctly requires understanding several related concepts: the distinction between a nitrosamine alert limit and an action limit, how to calculate acceptable intake when multiple nitrosamines are present in the same product, how less-than-lifetime (LTL) exposure calculations apply to short-duration therapies, and ultimately how those figures translate into defensible nitrosamine specification setting. A side-by-side comparison of nitrosamine AI limits across regulatory bodies is often the fastest way to spot where a global control strategy needs to satisfy the stricter of several regional values.
Key Difference 4: Analytical Testing Requirements
FDA places significantly greater emphasis on confirmatory laboratory testing than the general ICH M7 text. Testing generally involves highly sensitive analytical methods such as LC-MS/MS, LC-HRMS, GC-MS/MS, and GC-HRMS, often achieving detection limits in the low parts-per-billion or even parts-per-trillion range.
Building a testing program that can reliably hit these targets involves several technical decisions: choosing between direct injection and headspace techniques depending on nitrosamine volatility, achieving an ultra-low limit of quantitation (LOQ) appropriate to the AI limit in question, applying HRMS for nitrosamine testing when unknown or unexpected nitrosamines need to be identified, and overcoming matrix effects in LC-MS/MS for complex formulations. Every method also depends on properly qualified reference materials, which is why nitrosamine reference standard qualification is a foundational step before any confirmatory data can be considered reliable. The typical analytical workflow includes risk assessment, method development, method validation, confirmatory testing, data review, root cause investigation, CAPA implementation, and regulatory documentation — supported end-to-end by dedicated nitrosamine method development and validation services.
Key Difference 5: Root Cause Investigation
FDA guidance strongly emphasizes understanding exactly why nitrosamines form, rather than simply confirming that they are present. Potential sources include nitrite-containing raw materials, amines, solvent impurities, recycled solvents, process water, packaging interactions, manufacturing equipment, cleaning agents, degradation pathways, and environmental contamination.
Comprehensive root cause investigations — tracing findings back to specific nitrosamine formation pathways in API synthesis rather than stopping at the point of detection — support sustainable mitigation strategies rather than temporary corrective actions that leave the underlying chemistry unresolved.
Key Difference 6: Lifecycle Management
Both frameworks support lifecycle quality management, but FDA expects continuous reassessment whenever manufacturers change API suppliers, synthetic routes, manufacturing sites, equipment, packaging materials, storage conditions, excipients, or cleaning procedures. This is formalized through structured nitrosamine lifecycle management programs that treat nitrosamine control as an ongoing quality system activity rather than a one-time submission requirement.
Where confirmed findings exceed acceptable limits, sponsors typically need a documented nitrosamine reformulation strategy alongside broader nitrosamine control strategy development services to bring the product back into compliance without unnecessary disruption to supply.
6: Submission-Specific Requirements: NDA, ANDA, and Generic Products
Nitrosamine expectations also differ depending on the regulatory pathway. Understanding the distinctions in NDA vs. ANDA nitrosamine submission requirements is essential, since generic manufacturers face a somewhat different documentation burden than innovator sponsors. A properly scoped nitrosamine risk assessment for ANDA submissions needs to account for reference-listed drug history and prior FDA findings on the same molecule, which is a major part of what makes nitrosamine testing for generic drugs a distinct discipline from innovator-product testing.
Product- and Modality-Specific Nitrosamine Considerations
Because nitrosamine risk is highly chemistry- and formulation-dependent, FDA and ICH expectations play out differently across drug classes. Sponsors working on affected molecules or modalities benefit from targeted programs such as:
- Nitrosamine testing for rifampicin and nitrosamine testing for rifapentine, two rifamycin-class antibiotics with well-documented nitrosamine findings
- Nitrosamine testing for ranitidine, one of the earliest and most widely publicized nitrosamine recalls
- Nitrosamine testing for metformin, given its enormous prescription volume and multiple manufacturer recalls
- Nitrosamine testing for beta-blockers, a drug class with several confirmed NDSRI findings
- Nitrosamine testing for highly potent APIs, where containment and analytical sensitivity both add complexity
- Nitrosamine testing for high-risk drug classes more broadly, based on structural alert and manufacturing route
- Nitrosamine testing for OTC products, which carries added scrutiny given broad, unsupervised patient exposure
- Nitrosamine testing for injectable drug products, where formulation and container closure interactions add risk
- Nitrosamine testing in combination products and the broader question of nitrosamine risk in drug-drug combination products, where multiple actives can interact chemically
- Nitrosamine testing for veterinary drug products, an often-overlooked category subject to its own regulatory expectations
Stability, Batch Release, and Program Timelines
Nitrosamine control doesn’t end at initial risk assessment — it has to be built into ongoing quality operations. This includes nitrosamine testing in stability studies to confirm impurities don’t form or increase over shelf life, and clearly defined nitrosamine batch release testing requirements so that routine manufacturing doesn’t stall waiting on ad hoc testing decisions. Sponsors planning a compliance program should also map out a realistic nitrosamine testing timeline, since analytical method development and validation are frequently the longest lead-time items in the entire process.
7: Analytical Technologies Supporting Regulatory Compliance
Regulatory compliance depends heavily on reliable analytical testing across a range of platforms:
| Technology | Primary Application |
|---|---|
| LC-MS/MS | Routine nitrosamine quantification |
| LC-HRMS | Unknown nitrosamine identification |
| GC-MS/MS | Volatile nitrosamines |
| GC-HRMS | Ultra-trace volatile impurities |
| ICP-MS | Elemental impurity evaluation |
| Orbitrap HRMS | High-resolution impurity profiling |
| QTOF MS | Structural elucidation |
| LC-UV | Supporting impurity analysis |
Method validation should demonstrate specificity, accuracy, precision, sensitivity, robustness, linearity, recovery, and stability before any data is used to support a regulatory submission.
8: Best Practices for Regulatory Compliance
Organizations can strengthen compliance by adopting proactive quality systems rather than reactive testing. Recommended practices include:
- Conduct comprehensive nitrosamine risk assessments early in development, not just before filing.
- Evaluate all suppliers and raw materials for nitrosamine-forming potential.
- Monitor process changes using quality risk management principles.
- Use validated, ultra-sensitive analytical methods appropriate to the compound and matrix.
- Investigate all confirmed nitrosamine findings thoroughly, down to root cause.
- Establish scientifically justified control strategies rather than one-off corrective actions.
- Maintain complete documentation for inspections and regulatory queries.
- Perform periodic lifecycle reviews whenever suppliers, processes, or packaging change.
Common Industry Challenges
Despite clear regulatory guidance, many manufacturers encounter practical challenges, including ultra-low detection requirements, unknown nitrosamine structures, complex sample matrices, API-specific chemistry, multiple manufacturing sites, changing supplier networks, differing global regulatory expectations, and rapidly evolving toxicological data. Many organizations address these challenges by outsourcing nitrosamine testing to a specialized CRO rather than trying to build ultra-trace detection capability in-house, particularly for lower-volume or one-off testing needs.
9: How ResolveMass Laboratories Inc. Supports Nitrosamine Compliance
ResolveMass Laboratories Inc. provides comprehensive analytical support for pharmaceutical companies developing small molecules, biologics, and complex drug products. Its scientific capabilities include nitrosamine risk assessment, confirmatory testing, LC-MS/MS method development, HRMS characterization, unknown impurity identification, method validation, extractables and leachables (E&L) studies, forced degradation studies, regulatory-ready analytical reports, and CMC documentation support.
By combining advanced instrumentation with experienced scientific teams, ResolveMass helps sponsors generate reliable analytical data aligned with FDA, Health Canada, EMA, and ICH expectations — whether the work involves a single high-risk molecule or a portfolio-wide nitrosamine reassessment.
Conclusion:
Understanding ICH M7 vs FDA Nitrosamine Guidance is essential for pharmaceutical manufacturers navigating today’s increasingly stringent regulatory environment. While ICH M7 establishes the global scientific framework for controlling mutagenic impurities, FDA nitrosamine guidance expands those principles with detailed recommendations tailored specifically to nitrosamine risk assessment, analytical testing, root cause investigation, and lifecycle monitoring.
Organizations that integrate both frameworks into their pharmaceutical quality systems are better positioned to reduce regulatory risk, protect patient safety, and support successful global submissions. Partnering with an experienced analytical CRO such as ResolveMass Laboratories Inc. further strengthens compliance by providing scientifically rigorous testing, comprehensive risk assessments, and regulatory-ready documentation that meets evolving global expectations.
Frequently Asked Questions:’
Yes. FDA Nitrosamine Guidance is built upon the scientific principles established in ICH M7 for controlling mutagenic impurities. However, it provides additional nitrosamine-specific recommendations, including detailed risk assessment methodologies, analytical testing expectations, acceptable intake (AI) limits, and lifecycle management practices. While ICH M7 serves as a global framework, the FDA guidance expands these concepts to address the unique challenges associated with nitrosamine impurities in pharmaceutical products.
Although ICH M7 covers all DNA-reactive mutagenic impurities, the widespread discovery of nitrosamines in marketed medicines revealed the need for more detailed regulatory expectations. FDA guidance addresses specific nitrosamine formation pathways, testing requirements, root cause investigations, supplier evaluations, and ongoing monitoring that are not covered in the same level of detail within ICH M7. This helps manufacturers better manage nitrosamine-specific risks.
Acceptable Intake (AI) limits represent the maximum daily exposure to a nitrosamine impurity that is considered to present a negligible lifetime cancer risk. Regulatory agencies establish AI values using toxicological data and carcinogenic potency assessments. These limits vary for different nitrosamines and may be updated as new scientific evidence becomes available, making periodic regulatory review essential.
A nitrosamine risk assessment should be reviewed whenever significant changes occur in the manufacturing process, raw material suppliers, synthetic route, packaging materials, equipment, storage conditions, or cleaning procedures. Even without major changes, many companies incorporate periodic reviews into their pharmaceutical quality systems to ensure ongoing compliance with evolving regulatory guidance.
Nitrosamines typically form through reactions between nitrite-containing compounds and secondary or tertiary amines under suitable processing conditions. Factors such as contaminated raw materials, recycled solvents, process water, catalysts, excipients, degradation pathways, packaging interactions, and storage conditions can all contribute to nitrosamine formation if not properly controlled.
Yes. While nitrosamine concerns have primarily affected small-molecule drugs, biologics, peptides, and certain complex drug products may also require nitrosamine risk evaluations if their manufacturing processes, raw materials, excipients, or packaging systems present a potential risk for nitrosamine formation. A science-based assessment should determine whether further testing is necessary.
If nitrosamine levels exceed established AI limits, manufacturers should immediately initiate a comprehensive investigation to identify the root cause, evaluate patient risk, implement corrective and preventive actions (CAPA), perform confirmatory testing, and notify regulatory authorities when required. Additional process optimization and ongoing monitoring may also be necessary before product release.
Reference
- Khan HS, Despres-Gnis F, Stults CL, Mullis J, Nugara N, Sen A, Nagao L. An Overview and Discussion of N-Nitrosamine Considerations for Orally Inhaled Drug Products and Relevance to Other Dosage Forms. AAPS PharmSciTech. 2023 Jan 18;24(1):37.https://link.springer.com/article/10.1208/s12249-022-02491-7
- Patel MB, Usmani A. Evolution of nitrosamine regulations: Lessons from pharmaceuticals applied to food safety management.https://www.newresearchjournal.com/assets/archives/2025/vol10issue4/10083.pdf
- Umrethwala A, Mutalik R, Sharma S. Nitrosamine Impurities: Critical Insights into Chemistry, Analytical Tools, and Worldwide Regulatory Requirements. InAnnales Pharmaceutiques Françaises 2026 Apr 4. Elsevier Masson.https://www.sciencedirect.com/science/article/pii/S0003450926000441
- Yerram S, Muhammad Nizam VP, Srivastava S, Nanduri S. Nitrosamine Contamination in Pharmaceuticals: A Retrospective Regulatory Analysis of USFDA Recalls and Risk Mitigation Strategies (2018–2025). Therapeutic Innovation & Regulatory Science. 2026 Mar;60(2):519-33.https://link.springer.com/article/10.1007/s43441-025-00891-y
- Ramamoorthy S. Impurity Characterization Across Drug Development Stages: Analytical Methodologies and Regulatory Perspectives. Pharmaceutical Research. 2026 Mar 3:1-22.https://link.springer.com/article/10.1007/s11095-026-04054-y
- Jolly RA, Cornwell PD, Noteboom J, Sayyed FB, Thapa B, Buckley LA. Estimation of acceptable daily intake values based on modeling and in vivo mutagenicity of NDSRIs of fluoxetine, duloxetine and atomoxetine. Regulatory Toxicology and Pharmacology. 2024 Sep 1;152:105672.https://www.sciencedirect.com/science/article/pii/S0273230024001132

