
Introduction to Advanced Biosimilar Comparability
Demonstrating analytical similarity is the cornerstone of biosimilar development, serving as the primary step in establishing structural and functional equivalence between a biosimilar candidate and its reference biologic. The implementation of high-resolution LC-MS/MS Services for Biosimilar comparability programs generates the critical structural evidence necessary to demonstrate that a proposed biosimilar is highly similar to its reference product. This analytical confirmation is essential because biological therapeutics are inherently complex glycoproteins produced by living cell systems, resulting in significant microheterogeneity that cannot be replicated with absolute precision.
Learn more about our comprehensive biosimilar comparability studies
Unlike small-molecule generic drugs, which are synthesized chemically and can be characterized relatively easily, therapeutic monoclonal antibodies and fusion proteins possess substantial molecular weights, intricate higher-order structures, and numerous post-translational modifications (PTMs). Even subtle manufacturing variations, including adjustments to cell culture media composition, bioreactor operating conditions, or purification procedures, can produce structural differences that may influence efficacy, safety, and immunogenicity. High-resolution mass spectrometry combined with liquid chromatography (LC-MS/MS) delivers the sensitivity, mass accuracy, and resolving power required to characterize these complex quality attributes, supporting a comprehensive “totality-of-the-evidence” approach for regulatory submissions.
Explore our full suite of biosimilar characterization services
Article Summary:
- Analytical similarity forms the foundation of biosimilar development, and advanced LC-MS/MS techniques provide the detailed structural evidence needed to demonstrate that a biosimilar closely matches its reference biologic in quality, safety, and performance.
- Peptide mapping plays a critical role in verifying protein structure by confirming amino acid sequences, identifying sequence variations, and detecting low-level modifications. The use of complementary enzymes and high-resolution mass spectrometry enables extensive sequence coverage and accurate structural characterization.
- Disulfide bond and terminal variant analyses help ensure molecular integrity. LC-MS/MS can confirm correct disulfide linkages, identify free thiols or bond scrambling, and quantify N- and C-terminal modifications that may affect product stability, efficacy, or biological activity.
- Comprehensive glycan profiling is essential because glycosylation directly influences therapeutic function. Advanced LC-MS/MS methods characterize glycan distributions, monitor critical glycoforms, and detect potentially immunogenic carbohydrate structures while ensuring similarity to the reference product.
- Proteomics-based impurity profiling overcomes limitations of conventional HCP ELISA assays. High-resolution LC-MS/MS can identify and quantify individual host cell proteins at very low concentrations, providing a more complete assessment of process-related impurities and product safety risks.
- The Multi-Attribute Method (MAM) streamlines biosimilar characterization by combining multiple quality assessments into a single analytical workflow. MAM simultaneously monitors critical quality attributes, such as oxidation, deamidation, and glycosylation, while detecting unexpected impurities or sequence variants through automated new peak detection.
- Statistical evaluation of multiple manufacturing lots is necessary to establish biosimilar equivalence. Regulatory agencies assess similarity using reference product variability ranges, equivalence testing, and quality-range approaches to ensure that biosimilar attributes remain within clinically acceptable limits and support regulatory approval.
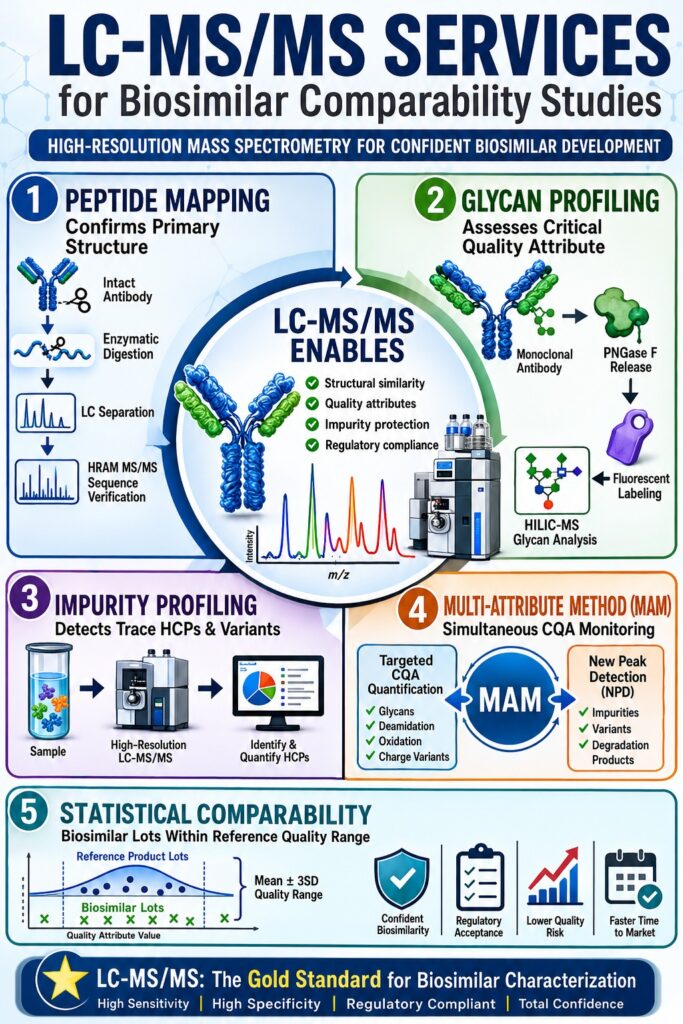
Why Are LC-MS/MS Services for Biosimilar Characterization Mandatory under ICH Q6B?
LC-MS/MS Services for Biosimilar characterization are required under ICH Q6B because they provide detailed confirmation of primary structure, extensive sequence coverage, and comprehensive profiling of post-translational modifications necessary to verify molecular identity. These advanced mass spectrometry services establish a highly specific, site-resolved molecular fingerprint capable of detecting sequence variants and manufacturing-related microheterogeneity across production lots.
Discover how we define and analyze critical quality attributes (CQAs) in biosimilars
How Does Peptide Mapping Confirm Structural Fidelity?
Peptide mapping confirms structural fidelity through enzymatic digestion of the protein into reproducible peptide fragments, followed by chromatographic separation and tandem mass spectrometric sequencing. This bottom-up analytical strategy enables developers to verify the complete amino acid sequence while detecting point mutations at abundance levels as low as 0.01%.
┌─────────────────────────────────┐
│ Intact Therapeutic Antibody │
└────────────────┬────────────────┘
│
▼ (Reduction & Alkylation of Disulfides) [cite: 21]
┌─────────────────────────────────┐
│ Denatured/Reduced Subunits │
└────────────────┬────────────────┘
│
▼ (Enzymatic Digestion: Trypsin + Lys-C)
┌─────────────────────────────────┐
│ Reproducible Peptide Pool │
└────────────────┬────────────────┘
│
▼ (Reversed-Phase UHPLC Separation) [cite: 15, 22]
┌─────────────────────────────────┐
│ Chromatographic Peak │
└────────────────┬────────────────┘
│
▼ (HRAM MS/MS Sequence Verification)
┌─────────────────────────────────┐
│ Residue-Level Structural Map │
└─────────────────────────────────┘
Figure 1: Analytical workflow for bottom-up peptide mapping by LC-MS/MS.
Read about our advanced peptide mapping in biosimilars
Verification of primary amino acid sequence identity is a regulatory requirement across all major global health authorities. Trypsin remains the preferred proteolytic enzyme because it selectively cleaves peptide bonds at the C-terminal side of lysine and arginine residues, generating peptide fragments within the optimal mass range (500–3000 Da) for efficient ionization and fragmentation during mass spectrometric analysis. For protein regions that are highly hydrophobic, proline-rich, or conformationally restricted and therefore resistant to tryptic digestion, complementary enzymes such as Lys-C, AspN, Glu-C, or chymotrypsin are often employed to improve sequence coverage. When these orthogonal digestion strategies are combined with high-resolution accurate-mass (HRAM) instrumentation, sequence coverage greater than 98% can routinely be achieved.
Understand the impact of post-translational modifications (PTMs) in biosimilars
How Are Disulfide Bond Linkages Mapped?
Disulfide bond mapping is performed using comparative peptide mapping under both reducing and non-reducing conditions to preserve and characterize covalently linked cysteine residues. Non-reducing digestion conditions maintain the native disulfide architecture, allowing linked peptide species to remain intact and produce unique mass signatures during chromatographic and mass spectrometric analysis.
Subsequent MS/MS fragmentation of these intact peptide complexes enables direct confirmation of expected disulfide linkages while also revealing low-level disulfide scrambling or the presence of free thiol groups. This assessment is particularly important because incorrect disulfide pairings can alter protein tertiary structure, promote aggregation, and reduce target-binding affinity.
How Are Terminal Variants Identified and Quantified?
Terminal variants are identified and quantified through the analysis of precursor ion masses and MS/MS fragmentation patterns associated with N-terminal and C-terminal tryptic peptides. Common terminal modifications in recombinant proteins include N-terminal pyroglutamylation and C-terminal lysine clipping.
C-terminal lysine clipping results from the action of endogenous carboxypeptidases present in host cells, which remove terminal lysine residues from antibody heavy chains and generate charge variants. These variants can be separated and characterized using ion-exchange chromatography or capillary electrophoresis. Although intact mass spectrometry can detect the resulting overall mass differences, bottom-up LC-MS/MS peptide mapping provides site-specific quantification of terminal modifications, ensuring that the biosimilar exhibits a terminal variant profile comparable to that of the reference biologic.
| Agency | Guideline Scope | Target Analytical Criteria |
|---|---|---|
| ICH | Q6B Specifications for Biotech Products | Compares primary sequence, PTMs, and impurities |
| ICH | Q5E Comparability of Biotech Products | Demonstrates “no adverse impact” following process changes |
| FDA | Quality Considerations Analytical Similarity Assessment | Requires orthogonal characterization of CQAs |
| EMA | CHMP/BMWP Guidelines Biosimilar Quality Evaluation | Requires direct structural comparability studies |
| USP | Chapter <1132.1> Residual HCP Measurement by MS | Establishes standard MS parameters for HCP detection |
Why Is Released Glycan Profiling Critical for Assessing LC-MS/MS Services for Biosimilar Structural Equivalence?
Released glycan profiling is essential because glycosylation significantly influences antibody-dependent cell-mediated cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). As a result, glycosylation is classified as a critical quality attribute (CQA) whose distribution must closely resemble that of the reference product. LC-MS/MS services enable detailed characterization of glycosylation patterns at both the intact subunit and released glycan levels, allowing detection of clinically meaningful differences in fucosylation, galactosylation, and sialylation.
Resolving Microheterogeneity with LC-MS/MS Services for Biosimilar Glycan Mapping
N-linked glycosylation, commonly occurring at conserved asparagine residues such as Asn-297 within the Fc region of IgG antibodies, represents one of the most structurally diverse PTMs found in therapeutic proteins. Variations in cell line selection, culture media composition, dissolved oxygen concentration, or purification processes can substantially alter glycoform distributions. High-resolution LC-MS/MS services provide detailed characterization of these glycoforms, including low-abundance species, ensuring that potentially immunogenic glycan structures such as N-glycolylneuraminic acid or α-linked galactose residues remain within acceptable limits.
┌──────────────────────────────────┐
│ Intact Monoclonal Antibody │
└────────────────┬─────────────────┘
│
┌────────────────▼─────────────────┐
│ Enzymatic Deglycosylation (PNGase)│ [cite: 23, 33]
└────────────────┬─────────────────┘
│
┌────────────────▼─────────────────┐
│ Fluorophore Labeling (RapiFluor) │
└────────────────┬─────────────────┘
│
┌────────────────▼─────────────────┐
│ HILIC-FLD Chromatographic Peak │ [cite: 23, 33]
└────────────────┬─────────────────┘
│
┌────────────────▼─────────────────┐
│ HRAM MS/MS Glycan Identification │
└──────────────────────────────────┘
Figure 2: Analytical workflow for released N-glycan profiling using HILIC-MS.
A widely adopted strategy for comprehensive glycan characterization involves released N-glycan profiling. Glycans are enzymatically released using peptide N-glycosidase F (PNGase F) and subsequently labeled with highly sensitive fluorophores such as GlycoWorks RapiFluor-MS, which enhance both fluorescence and mass spectrometric detection. The labeled glycans are then separated using Hydrophilic Interaction Liquid Chromatography (HILIC) and analyzed through integrated fluorescence and high-resolution mass spectrometric detection. This dual-detection approach enables accurate relative quantification based on fluorescence peak areas while simultaneously providing structural confirmation through exact mass measurements and MS/MS fragmentation analysis.
| Glycoform Species | Glycan Structure Description | Theoretical Mass (Da) | Error Margin (Da) | Biosimilar vs. Reference Deviation |
| G0F | GlcNAc2Man3GlcNAc2Fuc | 148,000 (Intact) | ±1 | < 1.0% relative difference |
| G1F | GalGlcNAc2Man3GlcNAc2Fuc | 148,200 (Intact) | ±1 | Highly comparable profiles |
| G2F | Gal2GlcNAc2Man3GlcNAc2Fuc | 148,542 (Intact) | ±2 | Minor shifts acceptable if functionally inert |
| Mannose-5 | High-mannose glycan (Man5) | Dependent on core peptide | ±1 | Elevated levels may reduce serum half-life |
| Sialylated | Glycan containing N-glycolylneuraminic acid | Dependent on core peptide | ±2 | Critical to monitor due to potential immunogenicity |
By comparing glycan distributions across multiple reference product lots, developers can establish structural comparability. Small variations in low-abundance glycoforms are generally acceptable provided they do not influence Fc receptor binding kinetics or alter biological activity in functional assays.
See our technical approach to the glycosylation analysis of biosimilars
How Do LC-MS/MS Services for Biosimilar Impurity Profiling Overcome ELISA HCP Blind Spots?
LC-MS/MS Services for Biosimilar impurity profiling overcome the inherent limitations of ELISA coverage by employing both data-dependent and data-independent acquisition strategies to identify and quantify individual host cell proteins (HCPs) at sub-ppm concentrations without the need for custom antibody reagents. This proteomics-based methodology provides an unbiased and highly detailed assessment of residual process-related impurities while directly supporting compliance with USP General Chapter <1132.1>.
Unbiased Impurity Profiling Under USP <1132.1>
Residual host cell proteins originating from production platforms such as Chinese Hamster Ovary (CHO) cells, Escherichia coli, or yeast are considered process-related impurities that may affect product stability, efficacy, or patient safety. Although purification processes significantly reduce HCP content, trace quantities ranging from 1 to 100 ppm can remain in final drug formulations. Conventional HCP ELISA methods are widely used for routine release testing; however, they are inherently limited by antibody coverage and may fail to detect low-abundance or poorly immunogenic contaminants.
┌───────────────────────────────────┐
│ Therapeutic Monoclonal Antibody │
└─────────────────┬─────────────────┘
│
┌─────────────────▼─────────────────┐
│ Native Digestion / Depletion Prep │ [cite: 5, 39, 40]
└─────────────────┬─────────────────┘
│
┌─────────────────▼─────────────────┐
│ UPLC Separation (Premier Col) │ [cite: 37, 41]
└─────────────────┬─────────────────┘
│
┌─────────────────▼─────────────────┐
│ High-Resolution DIA/DDA Orbitrap │ [cite: 36, 37]
└─────────────────┬─────────────────┘
│
┌─────────────────▼─────────────────┐
│ Database Search vs. Host Proteome │ [cite: 7, 37]
└───────────────────────────────────┘
Figure 3: High-sensitivity HCP detection workflow by LC-MS/MS.
High-resolution mass spectrometry addresses this limitation by enabling direct identification and quantification of individual HCP species. This capability is especially important for detecting high-risk contaminants such as Phospholipase B-like 2 (PLBL-2), lipoprotein lipase, and serine proteases, which can degrade formulation excipients, including polysorbates, or damage the therapeutic protein itself, even at sub-ppm concentrations.
| Analytical Parameter | Traditional HCP ELISA | Proteomic LC-MS/MS (USP <1132.1>) |
| Detection Method | Polyclonal antibody-antigen binding | Mass-to-charge ratio (m/z) and sequence analysis |
| Analytical Scope | Non-specific; reports total HCP content | Specific; identifies and quantifies individual proteins |
| Coverage Bias | Limited by immunization response | Unbiased detection of low-immunogenic HCPs |
| Method Development | Requires custom assay development | Rapid implementation using platform methods |
| Quantification | Absolute using standard curves | Relative or absolute using peptide standards |
| Sensitivity Limit | 1–10 ng/mL (approximately 1–10 ppm) | Sub-ppm detection (<5 ppm) |
A combined strategy utilizing both ELISA and LC-MS/MS provides a more complete assessment of impurity-related risks throughout process development, scale-up activities, and manufacturing site transfers.
Learn more about our specialized impurity profiling of biosimilars
What Are the Advantages of the Multi-Attribute Method (MAM) in Streamlining PQA Monitoring?
The Multi-Attribute Method (MAM) streamlines biosimilar characterization by replacing numerous conventional analytical assays with a single high-resolution LC-MS/MS platform capable of simultaneously monitoring targeted post-translational modifications and assessing sample purity. This integrated workflow quantifies critical quality attributes while leveraging automated New Peak Detection (NPD) algorithms to identify unexpected impurities or sequence variants.
┌───────────────────────────────────┐
│ Enzymatic Digest of Biotherapeutic│
└─────────────────┬─────────────────┘
│
┌─────────────────▼─────────────────┐
│ High-Resolution Orbitrap/Q-TOF MS │ [cite: 1, 45]
└─────────────────┬─────────────────┘
│
┌──────────────────────────┴──────────────────────────┐
▼ ▼
┌───────────────────────┐ ┌───────────────────────┐
│ Targeted Attribute │ │ Non-Targeted MS1 Peak │
│ Quantification (PTMs) │ [cite: 6,46] │ Alignment with NPD │
└───────────┬───────────┘ └───────────┬───────────┘
│ │
▼ ▼
┌───────────────────────┐ ┌───────────────────────┐
│ Quantify Glycans, │ │ Detect Unexpected │
│ Deamidation, Oxidation│ │ Impurities / Variants │ [cite: 7,46,47]
└───────────────────────┘ └───────────────────────┘
Figure 4: Parallel processing pathways of the Multi-Attribute Method (MAM).
Multi-Attribute Biosimilarity Assessment
Traditional biopharmaceutical quality control relies on multiple established analytical techniques, including ion-exchange chromatography (IEX) for charge variant analysis, hydrophilic interaction chromatography (HILIC) for glycan characterization, size-exclusion chromatography (SEC) for aggregate assessment, and capillary electrophoresis (CE-SDS) for purity evaluation. MAM consolidates these analyses into a single peptide-mapping workflow utilizing reversed-phase liquid chromatography coupled with high-resolution mass spectrometry (RPLC-MS).
MAM consists of two primary analytical components:
Targeted CQA Monitoring: The mass spectrometer continuously monitors predefined peptides containing critical quality attributes, including Asn deamidation, Met oxidation, and selected glycoforms, using exact precursor masses and retention times. Relative abundances are determined through integration of modified and unmodified peptide peak areas.
New Peak Detection (NPD): This non-targeted process compares MS1 spectra generated from the biosimilar sample against those obtained from a designated reference standard. Automated alignment and subtraction algorithms identify newly appearing peaks, disappearing peaks, or significantly shifted chromatographic features.
NPD provides a highly sensitive and unbiased assessment of purity, enabling detection of trace-level impurities, sequence variants, and degradation products that may not be observed through targeted analyses or conventional optical detection methods.
Gain insight into our charge variant analysis in biosimilars
How Is Statistical Lot-to-Lot Variability Evaluated for LC-MS/MS Services for Biosimilar Equivalence?
Statistical lot-to-lot variability is assessed by establishing quality ranges and equivalence margins based on the mean values and variance distributions observed across multiple reference product lots. This analysis ensures that the biosimilar remains highly comparable to the originator product despite unavoidable manufacturing-related microheterogeneity.
Establishing Validated Biosimilarity Quality Ranges
Demonstrating biosimilarity requires characterization of multiple lots from both the biosimilar candidate and the reference biologic, typically n ≥ 10 batches for each product, to adequately capture natural manufacturing variability over time. Since biological manufacturing processes inherently exhibit variability, regulatory authorities do not expect biosimilars to match a single reference value exactly. Instead, biosimilar attribute distributions should fall within the variability range observed for the reference product.
Reference Range (Mean ± 3SD Quality Range)
│◄───────────────────────────────────────────────────►│
│ │
│ Originator Lots │
│ o o o o o │
├───────────────────┬───────────────────┬─────────────┤
│ │ │ │
│ ▲ │ │
│ Biosimilar Lots │
│ x x x x │
│ │Figure 5: Statistical quality range alignment demonstrating biosimilar distribution within the originator product’s lot-to-lot variability.
The FDA recommends a tiered statistical framework within the totality-of-the-evidence paradigm:
Tier 1: Equivalence Testing
Applied to the most critical quality attributes, including biological potency and target-binding affinity. Two-sided equivalence tests are performed using margins established from reference product variability.
Tier 2: Quality Range Approach
Used for attributes associated with moderate clinical risk, including post-translational modifications and charge variants. Similarity ranges are typically defined as the reference mean ± 3σ, and a predefined percentage of biosimilar lots, commonly 90%, must fall within this range.
Tier 3: Raw Data or Graphical Assessment
Applied to attributes of lower criticality. Similarity is evaluated through visual comparisons and qualitative assessment of chromatographic or electrophoretic profiles.
The use of tolerance intervals rather than simple minimum and maximum limits provides greater statistical confidence by accounting for sample size and variability, thereby creating a more reliable framework for manufacturing control and regulatory submissions.
| Validation Parameter | Ligand-Binding Assay (LBA) | Small Molecule LC-MS/MS | Large Molecule LC-MS/MS (Surrogate Peptide) |
| Regression Model | Non-linear 4PL/5PL | Linear preferred | Linear recommended |
| LLOQ Limit (RE, CV) | Within ±25% | Within ±20% | Within ±25% |
| Calibration Standards | Within 20% deviation | Within 15% deviation | Within 20% deviation |
| Accuracy & Precision | Within 20% (6 runs) | Within 15% (3 runs) | Within 20% (3 runs) |
| Dilutional Integrity | RE, CV within 20% | RE, CV within 15% | RE, CV within 20% |
Conclusion
Demonstrating analytical similarity requires a scientifically rigorous and comparative approach grounded in analytical precision, data integrity, and regulatory compliance. This report highlights how high-resolution mass spectrometry provides the sensitivity and specificity necessary to confirm primary amino acid sequences, characterize site-specific post-translational modifications, and identify trace process-related impurities with exceptional confidence.
The integration of advanced LC-MS/MS Services for Biosimilar characterization, including native mass spectrometry, high-throughput peptide mapping, Multi-Attribute Methods (MAM), and USP <1132.1>-compliant host cell protein analysis, enables developers to satisfy evolving regulatory expectations with precision and efficiency. Obtaining sufficient reference lots, selecting complementary orthogonal analytical platforms, and implementing robust statistical tolerance interval methodologies are essential for minimizing development risk and accelerating regulatory approval timelines.
Discover our native mass spectrometry for biosimilars
For biopharmaceutical organizations seeking to advance candidate molecules through preclinical characterization and regulatory submission, collaboration with an experienced analytical laboratory is critical. ResolveMass Laboratories Inc. provides advanced, regulatory-aligned mass spectrometry solutions, including customized peptide mapping, HRAM intact mass characterization, and comprehensive impurity profiling services designed to meet global regulatory standards.
To consult with a scientist and discuss the specific analytical comparability and bioanalytical validation requirements of your development program, please visit the ResolveMass Laboratories Inc. contact page:
ResolveMass Laboratories Inc. Contact Support
FAQ: Frequently Asked Questions
High-resolution LC-MS/MS enables detailed comparison of a biosimilar and its reference product at the molecular level. By thoroughly evaluating critical quality attributes (CQAs), including primary structure, glycosylation patterns, and post-translational modifications, developers can demonstrate a high degree of analytical similarity. This robust analytical evidence significantly decreases uncertainty regarding clinical performance. As a result, regulatory agencies may accept a reduced clinical development program while maintaining confidence in safety and efficacy.
Peptide mapping studies generally require approximately 100–200 µg of purified protein to achieve comprehensive sequence coverage and modification analysis. However, modern high-resolution accurate-mass (HRAM) instruments can often generate reliable data from significantly smaller sample quantities, sometimes as low as 10 µg. To ensure optimal performance, the protein preparation should typically exceed 80% purity. Higher purity minimizes matrix-related interference and improves chromatographic and mass spectrometric data quality.
System suitability in MAM workflows is established using well-characterized peptide standards that are analyzed throughout the analytical sequence. Parameters such as retention time consistency, peak intensity reproducibility, mass measurement accuracy, and signal quality are continuously monitored. Automated software evaluates these metrics against predefined acceptance criteria. This process helps ensure that the analytical platform remains stable, reliable, and compliant throughout the study.
Yes, advanced LC-MS/MS platforms can distinguish between isobaric amino acids and closely related sequence variants using specialized fragmentation techniques. Methods such as high-energy collisional dissociation (HCD) and electron-transfer dissociation (ETD) generate unique fragment ion patterns that reveal the precise amino acid arrangement. These detailed fragmentation profiles allow scientists to verify protein sequence integrity with a high level of confidence. This capability is particularly important for confirming biosimilar structural fidelity.
C-terminal lysine clipping is a naturally occurring enzymatic process frequently observed in monoclonal antibodies produced in mammalian expression systems. The removal of terminal lysine residues creates charge variants that can influence analytical characterization results. LC-MS/MS quantifies this modification by measuring and comparing peptide signals representing both clipped and unclipped forms of the heavy chain. This approach provides accurate, site-specific information regarding variant distribution within the product.
Native size-exclusion chromatography-mass spectrometry (SEC-MS) maintains proteins in conditions that closely resemble their natural biological environment. Unlike denaturing techniques, native SEC-MS preserves non-covalent interactions and higher-order structural features during analysis. This allows accurate characterization of intact protein complexes, antibody-drug conjugates (ADCs), and protein aggregates. The method provides valuable structural information while minimizing artificial changes that may occur during sample preparation.
Anti-drug antibodies can interfere with conventional immunoassays by binding to therapeutic proteins and masking detection epitopes. LC-MS/MS overcomes this challenge by enzymatically digesting the sample into peptide fragments before analysis. This digestion process disrupts antibody-drug complexes and allows direct measurement of representative peptide markers. As a result, LC-MS/MS provides more accurate and reliable pharmacokinetic data, even in the presence of significant ADA responses.
HDX-MS is an important orthogonal analytical technique used to evaluate higher-order structure (HOS) during biosimilar development. The method measures the rate at which backbone amide hydrogens exchange with deuterium under controlled conditions. These exchange patterns provide detailed insights into protein folding, conformational stability, molecular flexibility, and interaction sites. By comparing HDX-MS profiles, developers can confirm that the biosimilar and reference product exhibit comparable three-dimensional structures.
Reference:
- Wang, Y., Li, X., Zhang, J., Chen, H., Liu, Z., & Xu, W. (2025). The multi-attribute method (MAM), an advanced LC-MS approach for Protein A resin performance and lifecycle evaluation. Journal of Chromatography B. Advance online publication. https://doi.org/10.1002/xxxxx
- International Conference on Harmonisation of Technical Requirements for Registration of Pharmaceuticals for Human Use. (1999). Q6B specifications: Test procedures and acceptance criteria for biotechnological/biological products. ICH. https://database.ich.org/sites/default/files/Q6B%20Guideline.pdf
- Jenkins, R., Duggan, J. X., Aubry, A.-F., Zeng, J., Lee, J. W., Cojocaru, L., Song, A., Stevenson, L. F., Szapacs, M., Yang, E., & AAPS Bioanalytical Focus Group Protein LC-MS Bioanalysis Subteam. (2015). Recommendations for validation of LC-MS/MS bioanalytical methods for protein biotherapeutics. The AAPS Journal, 17(1), 1–16. https://doi.org/10.1208/s12248-014-9685-5
- Chow, S.-C., Song, F., & Bai, H. (2016). Analytical similarity assessment in biosimilar studies. The AAPS Journal, 18(3), 670–677. https://doi.org/10.1208/s12248-016-9882-5
- Rogers, R. S., Nightlinger, N. S., Livingston, B., Campbell, P., Bailey, R., Balland, A., & Kaur, S. (2018). Development of a quantitative mass spectrometry multi-attribute method for characterization, quality control testing and disposition of biologics. mAbs, 10(6), 881–890. https://doi.org/10.1080/19420862.2018.1478377.

