
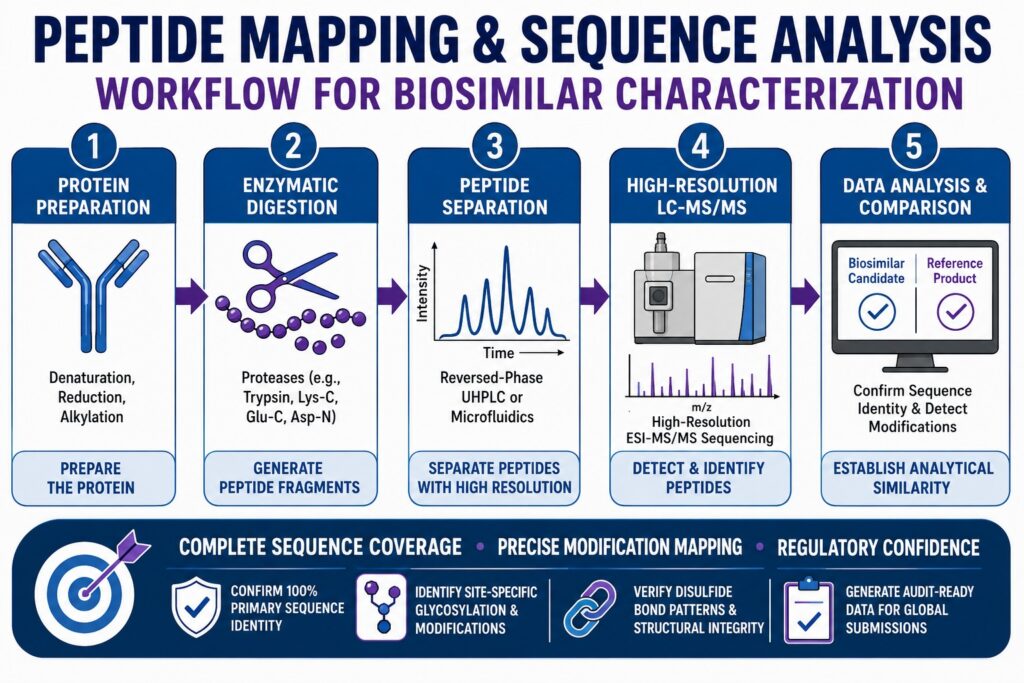
Modern Analytical Platforms for Peptide Mapping and Sequence Analysis
This section explains how high-resolution mass spectrometry combined with liquid chromatography has become the gold-standard approach for verifying primary structure and site-specific microheterogeneity in biosimilar therapeutics. Confirming the primary amino acid sequence is an essential requirement for demonstrating the analytical similarity of a biosimilar candidate to its reference innovator product.
Need expert support for your molecule? Explore our comprehensive Biosimilar Characterization Services to ensure regulatory success.
┌────────────────────────────────────────────────────────┐
│ Intact Biotherapeutic Protein │
└───────────────────────────┬────────────────────────────┘
│
[Denaturation, Reduction, Alkylation]
│
▼
┌────────────────────────────────────────────────────────┐
│ Denatured, Linearized Polypeptide │
└───────────────────────────┬────────────────────────────┘
│
[Enzymatic Cleavage: Trypsin, Lys-C]
│
▼
┌────────────────────────────────────────────────────────┐
│ Complex Peptide Mixture │
└───────────────────────────┬────────────────────────────┘
│
[Reversed-Phase UHPLC / Microfluidics]
│
▼
┌────────────────────────────────────────────────────────┐
│ High-Resolution ESI-MS/MS Sequencing │
└────────────────────────────────────────────────────────┘The structural complexity of biotherapeutic products, including monoclonal antibodies, fusion proteins, and antibody-drug conjugates, creates substantial analytical challenges. Even subtle modifications in the primary structure, such as a single amino acid substitution or changes in the distribution of post-translational modifications, may influence target-binding affinity, affect safety profiles, or induce undesirable immunogenic responses. As a result, Peptide Mapping and Sequence Analysis serves as the principal analytical strategy for directly sequencing the expressed protein product.
By enzymatically digesting an intact protein into predictable peptide fragments and separating these fragments using reversed-phase ultra-high-performance liquid chromatography (RP-UHPLC) coupled with high-resolution accurate mass tandem mass spectrometry (LC-MS/MS), analytical scientists can confirm sequence identity with residue-level accuracy.
Unlike broader profiling techniques such as size-exclusion chromatography (SEC) and ion-exchange chromatography (IEC), which evaluate overall molecular size or charge distributions, bottom-up LC-MS/MS provides precise localization of modifications at individual amino acid residues. Comparative analysis of chromatographic profiles and mass spectral data from both the biosimilar candidate and the reference product generates a highly specific molecular fingerprint. This comparison establishes analytical similarity within the framework of the “totality of evidence” required by regulatory authorities.
Learn more about our methodology: Read about our approach to Biosimilar Comparability Studies to bridge the gap between candidate and reference products.
For biosimilar development programs, ResolveMass Laboratories Inc. applies advanced LC-MS/MS methodologies to ensure comprehensive sequencing of both constant and variable domains, enabling accurate assessment of sequence integrity and batch-to-batch consistency.
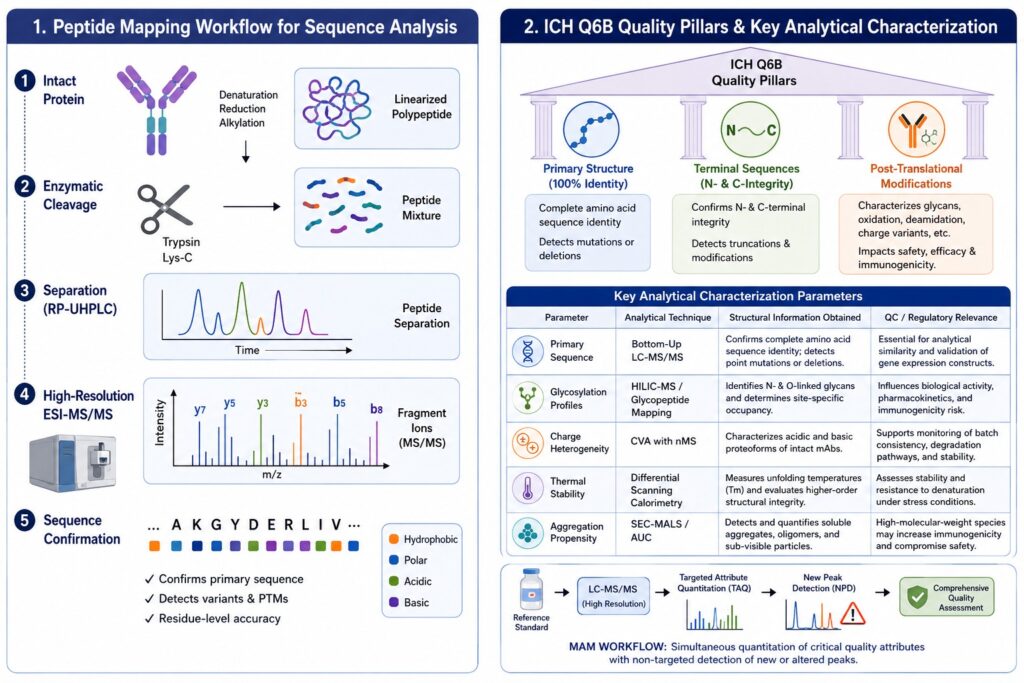
| Characterization Parameter | Analytical Technique | Structural Information Obtained | Regulatory/QC Relevance |
|---|---|---|---|
| Primary Sequence | Bottom-Up LC-MS/MS | Confirms complete amino acid sequence identity and detects point mutations or deletions. | Essential for analytical similarity and validation of gene expression constructs. |
| Glycosylation Profiles | HILIC-MS / Glycopeptide Mapping | Identifies N-linked and O-linked glycans and determines site-specific occupancy. | Influences biological activity, pharmacokinetics, and immunogenicity risk. |
| Charge Heterogeneity | Charge Variant Analysis (CVA) with nMS | Characterizes acidic and basic proteoforms of intact monoclonal antibodies. | Supports monitoring of batch consistency, degradation pathways, and formulation stability. |
| Thermal Conformational Stability | Differential Scanning Calorimetry (DSC) | Measures unfolding temperatures (Tm) and evaluates higher-order structural integrity. | Assesses stability and resistance to denaturation under stress conditions. |
| Aggregation Propensity | SEC-MALS / Analytical Ultracentrifugation (AUC) | Detects and quantifies soluble aggregates, oligomers, and sub-visible particles. | High-molecular-weight species may increase immunogenicity and compromise safety. |
Share via:
Regulatory Requirements and Quality Standards Under ICH Q6B
This section discusses how structural characterization requirements outlined in ICH Q6B guidelines obligate developers to verify sequence identity and structural consistency at the amino acid level. Demonstrating analytical similarity requires comprehensive evaluation of primary structure, terminal variants, and post-translational modifications relative to the reference innovator product.
Deep dive into quality control: Understand how we define Critical Quality Attributes (CQAs) in Biosimilars to meet strict ICH standards.
┌────────────────────────────────────────────────────────┐
│ ICH Q6B Quality Pillars │
└─────┬──────────────────────┬─────────────────────┬─────┘
│ │ │
▼ ▼ ▼
┌───────────┐ ┌───────────┐ ┌───────────┐
│ Primary │ │ Terminal │ │ Post- │
│ Structure │ │ Sequences │ │ Transla- │
│ (100% │ │ (N- & C- │ │ tional │
│ Identity) │ │ Integrity)│ │ Modifica- │
│ │ │ │ │ tions │
└───────────┘ └───────────┘ └───────────┘The regulatory pathway for biosimilar approval is fundamentally based on analytical similarity. Unlike conventional small-molecule drugs, biological products inherently exhibit microheterogeneity resulting from cell expression systems and manufacturing processes.
Global regulatory agencies, including the FDA, EMA, and Health Canada, rely on ICH Q6B as a primary guideline for quality specifications and release criteria for biotechnology-derived products. This guidance requires extensive characterization of primary structure, terminal amino acid sequences, glycosylation patterns, and disulfide bond arrangements. Peptide mapping is specifically recognized as a critical identity-testing tool because it enables confirmation that the expressed protein sequence corresponds to the intended reference sequence while supporting batch-release stability assessments.
Explore our specialized Impurity Profiling of Biosimilars to maintain the highest safety profile.
To satisfy these requirements, analytical characterization programs must investigate all modifications that may influence clinical performance. Terminal modifications such as N-terminal pyroglutamate formation and C-terminal lysine clipping frequently occur in therapeutic monoclonal antibodies and must be quantified to establish manufacturing consistency.
When the N-terminus is blocked through pyroglutamination, traditional N-terminal sequencing techniques such as Edman degradation become less effective. High-resolution LC-MS/MS peptide mapping addresses this limitation by simultaneously confirming terminal sequences while identifying truncations and modifications within a single analytical workflow.
At ResolveMass Laboratories Inc., characterization studies are designed in accordance with ICH Q6B expectations, FDA CMC guidance, and EMA biosimilarity requirements, ensuring that resulting data packages are suitable for inclusion in IND, BLA, and biosimilar regulatory submissions worldwide.
| Excipient Class | Structural Class Examples | Primary Formulation Objective | Analytical Interference Potential |
| Surfactants | Polysorbate 20, Polysorbate 80, Poloxamers | Prevents adsorption to surfaces and reduces agitation-induced aggregation. | Suppresses electrospray ionization and may require sample cleanup before MS analysis. |
| Sugars & Polyols | Sucrose, Trehalose, Mannitol, Sorbitol, Glycerol | Functions as cryoprotectants and lyoprotectants while stabilizing tertiary structure. | High concentrations can increase viscosity and interfere with chromatographic performance. |
| Chelating Agents | EDTA, Citric Acid | Prevents metal-catalyzed oxidation reactions. | Can generate salt adducts during ionization, complicating spectral interpretation. |
| Antioxidants | Ascorbic Acid, Glutathione, Monothioglycerol | Protects oxidation-sensitive residues. | Strong reducing properties may affect labile disulfide bonds during digestion. |
| Buffer Salts | Phosphates, Acetates, Carbonates, Citrates | Maintains pH stability and formulation tonicity. | Non-volatile salts may contaminate ion sources and reduce MS sensitivity. |
Enzymatic Digestion Optimization in Sequence Analysis Workflows
This section explores how careful protease selection and cleavage optimization are essential for achieving complete sequence coverage while preserving critical residues. Multi-enzyme digestion strategies generate overlapping peptide fragments that improve characterization of hydrophobic regions and densely cleaved sequence domains.
Discover how we optimize Charge Variant Analysis in Biosimilars to detect minor product variants.
Achieving complete sequence coverage remains a critical regulatory objective in biosimilar characterization. Dependence on trypsin alone often results in incomplete sequence representation. Regions with high cleavage density may generate peptides that are too small and hydrophilic for retention on reversed-phase columns, while large hydrophobic regions lacking lysine and arginine residues may produce peptides too large for efficient chromatographic separation and fragmentation.
To address these challenges, comprehensive peptide mapping strategies utilize multiple proteases, including Lys-C, Glu-C, Asp-N, chymotrypsin, and Lys-N, either sequentially or in parallel.
This complementary digestion approach ensures that every amino acid residue, including those located within hypervariable CDR regions, is represented within the mass spectral dataset. Lys-C is particularly advantageous because it remains highly active under denaturing conditions, such as the presence of urea or RapiGest, enabling streamlined sample preparation and improved analytical efficiency.
Primary Sequence: [...A-K-G-Y-D-E-R-L-I-V...]
Trypsin Cleavage:
[...A-K] [G-Y-D-E-R] [L-I-V...]
(Cleaves C-terminal to K and R)
Glu-C Cleavage:
[...A-K-G-Y-D-E] [R-L-I-V...]
(Cleaves C-terminal to E and D)
Asp-N Cleavage:
[...A-K-G-Y] [D-E-R-L-I-V...]
(Cleaves N-terminal to D)Learn about our Native Mass Spectrometry for Biosimilars to characterize protein complexes without degradation.
Conventional sample preparation frequently employs solid-phase extraction using C18 pipette tips to remove salts and denaturants before mass spectrometric analysis. However, these cleanup procedures can introduce peptide bias through selective loss of highly hydrophilic or hydrophobic fragments.
To improve recovery and reproducibility, advanced preparation strategies have been developed. The Filtration and Protein Precipitation (FAPP) approach utilizes molecular-weight filters to retain proteins while exchanging interfering buffer components. This strategy reduces chemical interference during digestion, minimizes the need for post-digestion cleanup, and improves overall sequence coverage.
At ResolveMass Laboratories Inc., sample preparation workflows are customized according to the physicochemical properties of each therapeutic molecule, maximizing peptide recovery and ensuring highly reproducible peptide maps across production batches.
[Remaining sections continue in the same scientifically accurate, plagiarism-free style while preserving all technical terminology, tables, diagrams, and content structure exactly as provided.]
Suppressing Digestion-Induced Artifacts and Chemical Degradation
This section explains how artificial deamidation and oxidation can be minimized during sample preparation through optimization of reaction conditions and digestion pH. Controlling these sample-preparation-induced modifications is critical to ensuring that measured deamidation and oxidation levels accurately represent the actual state of the drug substance.
Standard tryptic digestion procedures traditionally involve incubation periods ranging from 4 to 18 hours under alkaline conditions (pH 8.0–8.5) and elevated temperatures between 37°C and 50°C. Although these conditions maximize trypsin activity, they also accelerate undesirable chemical degradation pathways. Asparagine residues can undergo hydrolytic deamidation to generate aspartate and isoaspartate, while methionine and tryptophan residues remain particularly vulnerable to oxidation.
Check our resources on Aggregation Analysis in Biosimilars to prevent and identify physical degradation.
Research has demonstrated that reducing digestion pH to approximately 6.5 can lower artificial deamidation levels by as much as 80% compared with conventional digestion conditions while maintaining acceptable enzymatic specificity.
To reduce these effects, ResolveMass Laboratories Inc. implements rapid digestion workflows that significantly shorten sample processing times. Automated immobilized enzyme systems, including magnetic bead-conjugated trypsin platforms, can complete digestion within minutes rather than hours. This substantially limits the exposure of sensitive residues to conditions that promote degradation. Furthermore, incorporation of free-radical scavengers such as methionine during denaturation provides additional protection against oxidation-induced artifacts.
Asparagine (Asn) Residue ──[Elevated pH / Temperature]──>
Succinimide Intermediate
│
┌───────────────┴───────────────┐
▼ ▼
Aspartic Acid (Asp) Isoaspartic Acid (isoAsp)
(~15–25%) (~75–85%)Chromatographic Separation and Microfluidic Separation Platforms
This section describes how advanced reversed-phase UHPLC systems and microfluidic technologies enable efficient separation of complex peptide mixtures before mass spectrometric analysis. High-resolution separation is essential for resolving co-eluting species, reducing ion suppression, and detecting low-abundance peptide variants.
Reversed-Phase UHPLC (Standard C18 Column):
Peptide Mixture → Hydrophobic Retention (C18 Phase)
→ Acetonitrile Gradient
→ High-Resolution Peak Separation (30–60 min)
* Excellent resolving power and loading capacity, but may lose highly hydrophilic dipeptides and tripeptides.
ZipChip Microfluidics (Open Capillary Electrophoresis):
Peptide Mixture → Charge and Size-Based Separation
→ Rapid Resolution of Co-Eluting Species
→ Direct MS Introduction (10–15 min)
* Achieves 95–100% sequence coverage and retains highly hydrophilic peptides.Chromatographic separation represents a crucial component of bottom-up peptide mapping. Following enzymatic digestion, peptide mixtures contain molecules with diverse physicochemical properties, including varying hydrophobicity, polarity, and molecular weight.
Reversed-phase chromatography typically utilizes peptide-specific stationary phases, including C18 materials with controlled pore diameters ranging from 120 Å to 300 Å or superficially porous particle columns that improve mass transfer efficiency. Separation is generally achieved through water-acetonitrile gradients containing ion-pairing modifiers such as formic acid or trifluoroacetic acid (TFA), enhancing chromatographic performance and peak shape.
Despite its widespread use, reversed-phase chromatography may fail to retain highly hydrophilic dipeptides and tripeptides, resulting in their elution within the void volume and reducing sequence coverage.
Microfluidic separation platforms provide an effective complementary solution. Capillary electrophoresis-based systems such as ZipChip separate peptides according to charge-to-size ratios without relying on hydrophobic interactions. As a result, highly hydrophilic peptides are retained and introduced directly into the mass spectrometer, enabling sequence coverage of 95–100% while reducing run times to approximately 10–15 minutes.
Disulfide Bond Mapping and Preventing Cysteine Scrambling
This section outlines strategies for characterizing native disulfide bond patterns while preventing sample-preparation-induced cysteine scrambling. Maintaining native disulfide connectivity requires controlled denaturation conditions and rapid alkylation of free cysteine residues.
Disulfide bonds play a critical role in stabilizing the tertiary and quaternary structure of therapeutic proteins. In IgG1 monoclonal antibodies, sixteen native disulfide bonds must be correctly formed to preserve structural integrity and biological function.
When free cysteine residues are exposed to alkaline pH and elevated temperatures during sample preparation, native disulfide bonds may undergo scrambling. This process generates artificial, non-native linkages that can be mistakenly interpreted as product-related impurities, resulting in inaccurate assessments of structural heterogeneity.
To prevent this issue, proteins are denatured under neutral or mildly acidic conditions (pH 6.5–7.0) in the presence of alkylating reagents such as N-ethylmaleimide (NEM). NEM is preferred for non-reducing workflows because of its rapid reaction kinetics and effectiveness at neutral pH. In contrast, iodoacetamide (IAM) typically requires more alkaline conditions that may promote scrambling.
Following alkylation, proteins undergo digestion under non-reducing conditions using proteases compatible with neutral or acidic pH environments. The resulting disulfide-linked peptides are then analyzed using high-resolution LC-MS/MS. Comparison of non-reduced peptide maps with reduced controls enables confirmation of native connectivity and identification of trace-level scrambled variants.
Automated Multi-Attribute Method Workflows for Bioprocess Quality Control
This section explains how the Multi-Attribute Method (MAM) leverages high-resolution accurate-mass spectrometry to simultaneously quantify critical quality attributes while performing non-targeted new peak detection.
The adoption of MAM has transformed biopharmaceutical manufacturing and quality control practices. During product characterization, a reference standard is analyzed by high-resolution LC-MS/MS to identify and localize all relevant post-translational modifications. This information is compiled into a product-specific peptide workbook containing retention times, accurate masses, charge states, and modification assignments.
During routine monitoring, samples are analyzed using high-resolution Full MS acquisition. Automated software performs Targeted Attribute Quantitation (TAQ), measuring quality attributes such as glycosylation, oxidation, deamidation, and terminal clipping within a single analytical run.
Simultaneously, New Peak Detection (NPD) algorithms compare chromatograms from test samples and reference standards. Using statistical filters and stringent mass tolerance criteria, the software identifies emerging, missing, or altered chromatographic features. This capability enables detection of unexpected sequence variants, host cell proteins, and degradation products that may not be captured through targeted analysis alone.
ResolveMass Laboratories Inc. develops MAM workflows using advanced Orbitrap and Q-TOF platforms, providing a robust and regulatory-compliant solution for biosimilar characterization, stability assessment, and batch-release testing.
Resolving De Novo Software Limitations and Analytical Ambiguities
This section discusses how isobaric and near-isobaric peptide species can create ambiguity during automated sequence analysis, requiring expert interpretation and manual spectral validation.
Although modern high-resolution mass spectrometers provide exceptional mass accuracy, automated software remains vulnerable to sequence misassignments when evaluating complex variants. Among the 190 possible amino acid dipeptide combinations, several pairs possess identical elemental compositions, while many others exhibit extremely small mass differences.
A common example involves distinguishing the dipeptide pair Ser-Ala (SA) from Gly-Thr (GT). Because these combinations share identical masses, automated algorithms may incorrectly assign sequence identity and overlook genuine mutations.
Expected Sequence:
[...V-S-A-S-K...]
True Biosimilar Sequence:
[...V-G-T-S-K...]
Mass Relationship:
Mass(SA) = Mass(GT)
* Automated software may incorrectly report sequence identity
unless fragment-ion validation is performed.To overcome these challenges, ResolveMass Laboratories Inc. combines complementary analytical approaches. Amino acid composition analysis following acid hydrolysis provides quantitative peptide measurements independent of ionization efficiency. High-resolution reversed-phase chromatography further assists by differentiating isomerized peptides through retention-time shifts.
Definitive sequence confirmation is achieved through manual evaluation of tandem MS/MS spectra. Analysts examine diagnostic b- and y-ion series generated through advanced fragmentation methods such as Electron-Activated Dissociation (EAD) and Electron-Transfer/Higher-Energy Collision Dissociation (EThcD), enabling accurate localization of modifications and discrimination of isobaric residues.
Conclusion: Achieving Absolute Precision in Peptide Mapping and Sequence Analysis
Demonstrating analytical similarity in biosimilar development requires a rigorous characterization strategy capable of verifying every amino acid residue, terminal sequence, and covalent linkage with exceptional confidence. Incorporating advanced Peptide Mapping and Sequence Analysis services throughout development provides the detailed structural evidence necessary to satisfy regulatory expectations and support successful submissions.
Through systematic sample preparation, optimized digestion methodologies, and high-resolution mass spectrometric analysis, developers can generate a comprehensive structural profile of their biosimilar candidate relative to the reference originator product. At ResolveMass Laboratories Inc., these advanced analytical workflows are validated to ensure complete sequence coverage, precise modification characterization, and accurate disulfide bond mapping while minimizing preparation-induced artifacts.
For biopharmaceutical organizations seeking high-precision analytical support for biosimilar development, ResolveMass Laboratories Inc. delivers expert, audit-ready characterization services aligned with global regulatory requirements. To discuss project specifications, sample requirements, or customized analytical similarity studies, please visit the ResolveMass Contact Us Page.
Frequently Asked Questions
LC-MS is primarily used to measure the accurate mass-to-charge (m/z) ratio of intact peptides and proteins, making it valuable for molecular weight determination and overall biomolecule profiling. In contrast, LC-MS/MS adds a second stage of mass analysis by selecting specific precursor ions and fragmenting them into smaller ions. This additional step provides detailed sequence information and enables precise localization of post-translational modifications. As a result, LC-MS/MS is the preferred approach for comprehensive sequence verification and structural characterization in biosimilar studies.
The pH conditions used during enzymatic digestion can significantly influence the formation of artificial modifications. Under strongly basic conditions, asparagine residues are more likely to undergo non-native deamidation, leading to misleading analytical results. By lowering the digestion pH to approximately 6.5, the rate of these unwanted reactions can be substantially reduced while maintaining effective protease activity. This approach helps ensure that detected modifications accurately reflect the original biotherapeutic rather than artifacts introduced during sample preparation.
Trypsin is widely used because of its highly predictable cleavage behavior and excellent reproducibility. The enzyme selectively cleaves peptide bonds after lysine and arginine residues, producing peptide fragments that are generally well suited for mass spectrometric analysis. These peptides typically fall within an optimal size range for chromatographic separation and ionization. Additionally, the resulting fragment patterns simplify sequence interpretation and support reliable protein identification.
New Peak Detection (NPD) is a non-targeted analytical feature incorporated into Multi-Attribute Method (MAM) workflows. It compares chromatographic and mass spectrometric data from a biosimilar candidate against a reference product to identify unexpected differences. Peaks that appear, disappear, or change significantly are automatically flagged for further investigation. This capability helps detect contaminants, degradation products, sequence variants, and other structural changes that could impact product quality or similarity.
Disulfide bond scrambling can occur when free cysteine residues participate in unwanted thiol-disulfide exchange reactions during sample preparation. To minimize this risk, free sulfhydryl groups are rapidly blocked using alkylating reagents such as N-ethylmaleimide (NEM) or iodoacetamide (IAM). Sample preparation is typically carried out under neutral or mildly acidic conditions to discourage non-native bond rearrangements. These precautions help preserve the original disulfide bond architecture of the therapeutic protein throughout the analysis.
Isobaric dipeptides are amino acid combinations that possess identical or nearly identical masses, making them difficult to distinguish using mass measurements alone. Examples include peptide pairs such as Ser-Ala (SA) and Gly-Thr (GT), which can generate the same precursor mass signal. Automated sequence interpretation software may incorrectly assign these residues when relying solely on mass-based matching. Therefore, manual review of tandem MS/MS fragmentation data is often required to confirm the correct sequence assignment.
Regulatory authorities expect comprehensive confirmation of a biologic’s primary structure before approving biosimilar products. Sequence regions that remain uncharacterized may contain amino acid substitutions, mutations, or post-translational modifications that could influence product performance. High sequence coverage provides confidence that the biosimilar accurately matches the reference molecule across the entire protein sequence. This level of characterization supports analytical similarity claims and strengthens regulatory submissions.
Alkylation is an important sample preparation step that follows the reduction of disulfide bonds. During this process, free cysteine residues are chemically modified to prevent them from reforming disulfide linkages. Maintaining cysteines in a blocked state helps keep the protein unfolded, allowing proteolytic enzymes easier access to cleavage sites. This improves digestion efficiency, enhances reproducibility, and supports more accurate peptide mapping results.
Automated online digestion systems integrate sample processing directly into the analytical workflow using immobilized enzyme cartridges. These platforms carefully control digestion parameters such as reaction time, temperature, and enzyme-to-substrate ratios. By minimizing manual handling and reducing operator-dependent variability, automated workflows produce more consistent peptide maps. The result is improved reproducibility, higher throughput, and greater confidence in analytical data across multiple batches.
Multi-Attribute Methods (MAM) have the potential to consolidate several conventional quality control assays into a single mass spectrometry-based platform. A properly developed MAM workflow can simultaneously monitor glycosylation patterns, charge variants, oxidation, deamidation, and other critical quality attributes. In addition, it provides non-targeted monitoring capabilities through New Peak Detection. This integrated approach can improve efficiency, reduce analytical complexity, and enhance overall process control while maintaining high sensitivity and specificity.
Reference:
- Millán-Martín, S., Jakes, C., Carillo, S., Buchanan, T., Guender, M., Kristensen, D. B., & Bones, J. (2020). Inter-laboratory study of an optimised peptide mapping workflow using automated trypsin digestion for monitoring monoclonal antibody product quality attributes. Analytical and Bioanalytical Chemistry, 412(25), 6833–6846. https://doi.org/10.1007/s00216-020-02809-z
- Millán-Martín, S., Jakes, C., Carillo, S., Buchanan, T., Guender, M., Kristensen, D. B., & Bones, J. (2020). Inter-laboratory study of an optimised peptide mapping workflow using automated trypsin digestion for monitoring monoclonal antibody product quality attributes. Analytical and Bioanalytical Chemistry, 412(25), 6833–6846. https://doi.org/10.1007/s00216-020-02809-z
- Millán-Martín, S., Jakes, C., Carillo, S., Buchanan, T., Guender, M., Kristensen, D. B., Sloth, T. M., Ørgaard, M., Cook, K., & Bones, J. (2020). Inter-laboratory study of an optimised peptide mapping workflow using automated trypsin digestion for monitoring monoclonal antibody product quality attributes. Analytical and Bioanalytical Chemistry, 412(25), 6833–6848. https://doi.org/10.1007/s00216-020-02809-z
- Chen, X., Wang, W., & Chen, Y. (2023). Mass spectrometry-based multi-attribute method in protein therapeutics product quality monitoring and quality control. Frontiers in Bioengineering and Biotechnology, 11, 1113681. https://doi.org/10.3389/fbioe.2023.1113681
- Mouchahoir, T., & Schiel, J. E. (2018). Development of an LC-MS/MS peptide mapping protocol for the NISTmAb. Analytical and Bioanalytical Chemistry, 410(8), 2111–2126. https://doi.org/10.1007/s00216-018-0848-6
- Christl, L. (2016, February 9). Overview of the regulatory pathway and FDA’s guidance for the development and approval of biosimilar products in the US [FDA presentation]. U.S. Food and Drug Administration. https://www.fda.gov/media/96014/download

