
Introduction
Post-Approval Changes for Approved Biosimilars require a structured, science-driven comparability assessment conducted in accordance with the ICH Q5E framework to demonstrate that manufacturing modifications do not negatively influence product quality, safety, or clinical efficacy. This analytical evaluation must confirm that the biosimilar manufactured after the change remains highly comparable to the product manufactured before the change, thereby ensuring that its clinical performance continues to be consistent and predictable.
Learn more about our professional biosimilar characterization services for your next project.
Managing the lifecycle of therapeutic biologics is both a scientific and operational challenge, particularly when implementing Post-Approval Changes for Approved Biosimilars. Manufacturing processes for biological products are highly sensitive to factors such as environmental conditions, host cell expression behavior, and downstream purification parameters. Since the manufacturing process itself defines the product, modifications including process scale-up, changes in cell lines, or optimization of purification steps can introduce subtle variations in critical quality attributes (CQAs), including glycan distribution patterns and charge heterogeneity. Comprehensive analytical characterization services, such as those offered by ResolveMass Laboratories Inc., help minimize these lifecycle risks by generating highly sensitive orthogonal mass spectrometry and biophysical datasets that demonstrate structural consistency and biological comparability before regulatory submission. Recent advancements in regulatory expectations—particularly the U.S. Food and Drug Administration (FDA) Draft Guidance issued in October 2025 and Health Canada’s revised guidance released in May 2026—have significantly transformed the global regulatory landscape by replacing routine Phase III confirmatory clinical trials with highly sensitive analytical comparability-based approaches.
Read about the latest biosimilar comparability studies and how we can support your regulatory submissions.
Article Summary:
- Post-approval manufacturing changes for approved biosimilars must comply with ICH Q5E comparability principles, ensuring that any process modification does not adversely affect product quality, safety, potency, or clinical performance.
- Manufacturing changes are categorized by regulatory risk into major, moderate, and minor changes. The level of risk determines the type of regulatory submission and the amount of scientific evidence required to support the modification.
- Analytical comparability is the cornerstone of post-approval assessment. Manufacturers use orthogonal analytical techniques to evaluate critical quality attributes, including primary structure, higher-order structure, glycosylation, purity, aggregation, charge variants, and biological activity.
- A three-arm comparability strategy—comparing the post-change biosimilar with the pre-change product and the originator reference—helps detect biological drift and ensures long-term manufacturing consistency, particularly for significant process changes.
- Critical quality attributes are assessed using a risk-based tiered approach. High-risk attributes require rigorous statistical equivalence testing, medium-risk attributes are evaluated against predefined quality ranges, and low-risk attributes are confirmed through qualitative or trend-based assessments.
- Real-world biosimilar case studies demonstrate that substantial manufacturing improvements, such as cell line transitions and process optimization, can be successfully implemented using robust analytical characterization supported by targeted pharmacokinetic studies instead of repeating large clinical efficacy trials.
- Global regulatory agencies, including the FDA, Health Canada, and EMA, are increasingly prioritizing advanced analytical comparability over routine Phase III clinical efficacy studies, enabling more efficient lifecycle management while maintaining confidence in biosimilar quality, safety, and therapeutic performance.
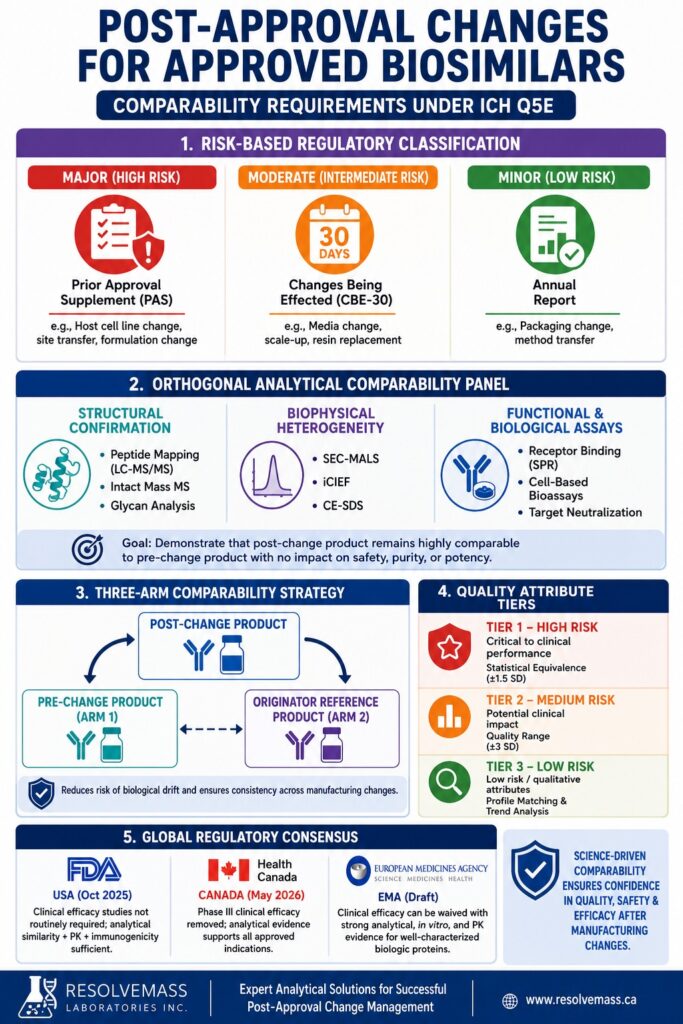
The Regulatory Paradigm for Post-Approval Changes for Approved Biosimilars
Regulatory pathways governing Post-Approval Changes for Approved Biosimilars follow a risk-based classification system established under the ICH Q5E and ICH Q12 guidelines. Within this framework, manufacturing modifications are categorized as major, moderate, or minor according to their potential impact on the quality, safety, and efficacy of the drug substance or finished drug product.
Consult our comprehensive guide on ICH Q6B guidelines for biological characterisation to align your quality control.
According to established regulatory frameworks, including the FDA’s 21 CFR 601.12, the European Medicines Agency (EMA) variation regulations, and Health Canada’s Food and Drug Regulations, manufacturers are required to thoroughly assess and document the consequences of manufacturing changes. Major modifications, which carry a substantial possibility of affecting product quality, require submission and regulatory approval through a Prior Approval Supplement (PAS). Moderate-risk changes are submitted as a Changes Being Effected in 30 Days (CBE-30) supplement, whereas minor modifications associated with minimal risk are documented within the annual report.
| Change Category | Potential Risk Level | Regulatory Submission Type | Representative Examples | Required Technical Documentation |
|---|---|---|---|---|
| Major | High | Prior Approval Supplement (PAS) | Host cell line or clone transition, manufacturing site transfer, formulation modification | Comprehensive analytical, structural, functional, stability, and, where appropriate, nonclinical or clinical PK bridging studies |
| Moderate | Intermediate | Changes Being Effected (CBE-30) | Media or feed modifications, manufacturing scale expansion, purification resin replacement | Comparative batch release testing, accelerated and stress stability studies, along with functional cell-based assays |
| Minor | Low | Annual Report | Secondary packaging supplier changes, analytical method transfer, raw material testing site modifications | Routine quality control release testing supported by historical process trend analysis |
Importantly, once a biosimilar receives marketing authorization, it is maintained under its own independent Biologics License Application (BLA). As a result, post-approval manufacturing changes are generally assessed by comparing the post-change biosimilar directly with the corresponding pre-change biosimilar using a qualified internal reference standard. Repeat head-to-head analytical comparisons with the original reference biologic are typically unnecessary unless specific changes in critical quality attributes indicate that additional evaluation is warranted.
Understand the nuances of a comparability exercise in biosimilar development to streamline your post-approval process.
Analytical Comparability Requirements Under the ICH Q5E Framework
The analytical comparability principles outlined in ICH Q5E require manufacturers to demonstrate that the biological product produced after a manufacturing change remains structurally and functionally highly comparable to the product manufactured before the change. This comprehensive analytical assessment must establish that any observed differences in quality attributes do not adversely influence the product’s safety, purity, or biological potency.
Establishing comparability does not require the post-change product to be identical to the pre-change product in every measured attribute. Instead, the objective is to demonstrate that any differences remain within scientifically justified and prospectively established acceptance ranges supported by historical manufacturing variability. Achieving this level of assurance requires the application of multiple orthogonal and complementary analytical technologies capable of evaluating every critical aspect of protein structure and function.

Through advanced analytical characterization platforms, including high-resolution mass spectrometry and multi-angle light scattering technologies, laboratories can detect even subtle post-translational modifications (PTMs) as well as emerging degradation pathways. Under the ICH Q5E framework, analytical comparability assessments are expected to comprehensively evaluate several fundamental quality pillars.
Check our forced degradation of biosimilars services to proactively identify stability issues.
Physicochemical Properties and Primary Structure
Manufacturers must demonstrate that the primary amino acid sequence remains unchanged between the pre-change and post-change materials by performing liquid chromatography–tandem mass spectrometry (LC-MS/MS) peptide mapping. In addition, terminal sequence variants, including C-terminal lysine clipping and N-terminal pyroglutamate formation, should be accurately quantified to verify sequence integrity and molecular homogeneity.
Higher-Order Structure (HOS) and Conformation
Confirmation of secondary and tertiary protein structure requires the use of multiple orthogonal biophysical characterization techniques. Commonly employed methods include Far-UV and Near-UV Circular Dichroism (CD) spectroscopy to evaluate secondary and tertiary structural organization, Differential Scanning Calorimetry (DSC) to determine thermal stability (Tm), and high-resolution Nuclear Magnetic Resonance (NMR) spectroscopy for detailed conformational analysis.
Purity, Impurities, and Size/Charge Heterogeneity
Size-Exclusion Chromatography coupled with Multi-Angle Light Scattering (SEC-MALS) serves as the standard analytical approach for detecting soluble protein aggregates, which are recognized as potential contributors to immunogenicity. Distribution of charge variants—including acidic, main, and basic species—should be characterized using cation-exchange chromatography (CEX-HPLC) or imaged capillary isoelectric focusing (iCIEF) to verify charge profile consistency following manufacturing changes.
Learn more about our precise aggregation analysis in biosimilars to minimize clinical risk.
Glycosylation and Post-Translational Modifications
Glycan composition, including levels of fucosylation, galactosylation, and high-mannose glycans, plays a critical role in influencing pharmacokinetic half-life as well as Fc-mediated biological functions. In addition, patterns of sialylation and overall glycan distribution are routinely characterized using Hydrophilic Interaction Liquid Chromatography with fluorescence detection (HILIC-FLD) or Capillary Electrophoresis to confirm that glycosylation profiles remain consistent after manufacturing modifications.
Discover how native mass spectrometry for biosimilars provides deeper structural insights
Mitigating Biological Drift: The Three-Arm Comparability Strategy
A three-arm, tiered comparability strategy plays a crucial role in minimizing the risk of biological drift by comparing the post-change biosimilar not only with its pre-change version but also with the originator reference product. This comprehensive approach helps ensure that successive manufacturing modifications do not gradually move the biosimilar beyond the structural and functional boundaries originally established by the innovator product.
For routine post-approval manufacturing changes, a direct comparison between the pre-change and post-change biosimilar is generally considered adequate. However, substantial process modifications carry a greater risk of introducing gradual cumulative differences, commonly referred to as manufacturing drift, which may not be detected through a simple two-way comparison. Implementing a three-arm comparability strategy enables sponsors to build a robust, multi-lot analytical database using the originator reference product, allowing scientifically justified, risk-based acceptance criteria to be established for evaluating manufacturing consistency.
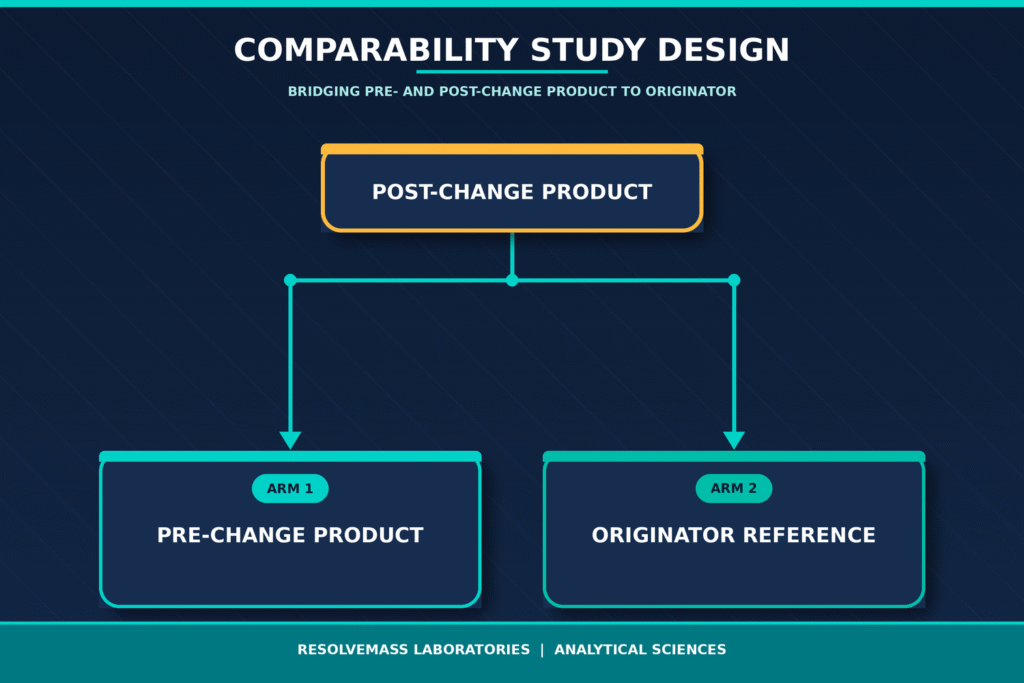
The analytical findings generated across these three comparison arms are typically organized into three statistical tiers according to the level of risk associated with each quality attribute.
Tier 1: High-Risk Attributes
Tier 1 includes critical quality attributes that have a direct influence on clinical performance, including target-binding potency and in vitro cell-based biological activity. According to FDA recommendations, these high-risk parameters are assessed using formal statistical equivalence testing. The objective is to demonstrate that the mean value of the post-change product remains within a predefined equivalence margin established from the historical variability of either the pre-change biosimilar or the innovator reference product.
θ1 ≤ (μpost − μpre) / σ ≤ θ2
In this equation, σ represents the standard deviation calculated from the historical analytical dataset, while the equivalence limits θ₁ and θ₂ are generally defined as ±1.5 × σ.
Tier 2: Medium-Risk Attributes
Tier 2 includes attributes that may have measurable or potential clinical relevance, such as glycan composition, including fucosylation and galactosylation percentages, as well as major charge variants. These parameters are evaluated using a Quality Range approach, where the analytical results for the post-change product are expected to remain within a ±3 SD interval established from the historical pre-change or reference product dataset.
Tier 3: Low-Risk Attributes
Tier 3 encompasses lower-risk characteristics or attributes that are primarily evaluated through qualitative assessment. Examples include confirmation of the primary amino acid sequence, higher-order structural integrity, and the presence of low-level impurities. Acceptance for these attributes is generally based on qualitative profile matching, historical trend analysis, or agreement with established theoretical physicochemical properties.
Explore the use of our advanced proteomics approach for biosimilars to monitor manufacturing consistency.
Post-Approval Changes for Approved Biosimilars: Case Studies in High-Risk Process Variations
Real-world examples of Post-Approval Changes for Approved Biosimilars provide valuable scientific evidence demonstrating how manufacturers successfully implement risk-based Quality-by-Design (QbD) strategies to support major manufacturing modifications while avoiding the need for repeated comparative clinical efficacy studies.
Case Study 1: Host Cell Line Transition in Bevacizumab Biosimilar (IBI305)
The post-approval manufacturing optimization of the bevacizumab biosimilar IBI305 represents one of the most technically challenging manufacturing changes reported to date. In this case, the manufacturer replaced a low-expression Chinese Hamster Ovary (CHO-K1S) production cell line with a high-expression CHO-K1SV GS-KO cell line, leading to an approximate threefold increase in production yield.
Because a host cell line transition is categorized as a major manufacturing change under the ICH Q5E guideline, the sponsor implemented a comprehensive three-arm analytical comparability program. The evaluation included multiple production batches of the post-change biosimilar, corresponding pre-change batches, and twenty-two commercial lots of the licensed reference product (Avastin) collected over a five-year period.
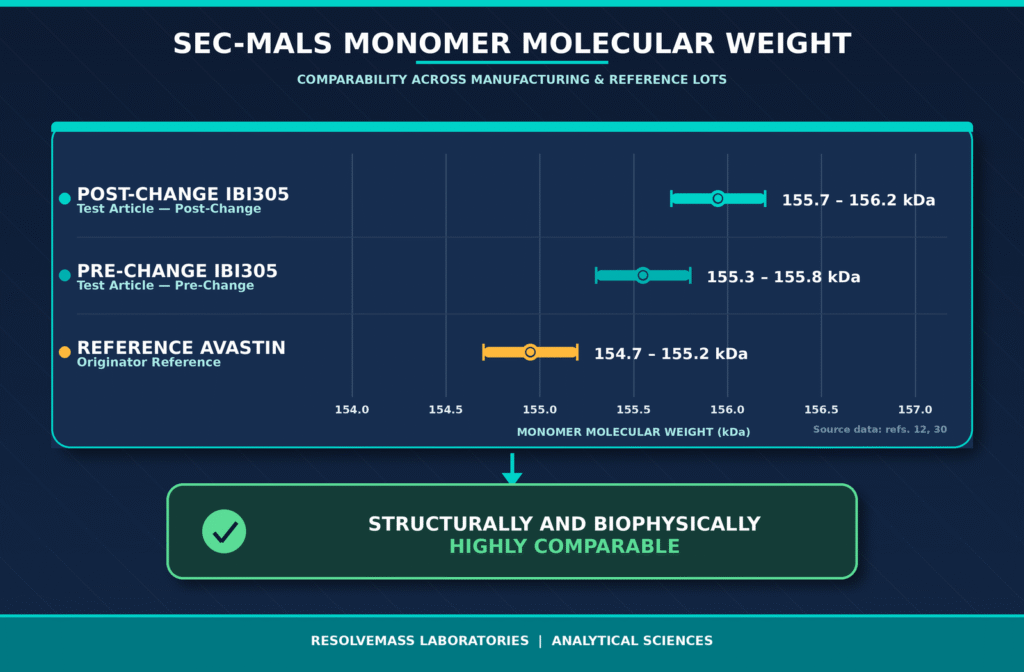
To eliminate any remaining uncertainty regarding clinical performance, the sponsor complemented the analytical comparability package with both nonclinical and clinical pharmacokinetic (PK) bridging studies. Demonstration of equivalent pharmacokinetics, pharmacodynamic activity, toxicological profile, and immunogenicity provided sufficient scientific evidence to support regulatory approval of the manufacturing change without requiring a comparative clinical efficacy trial.
Case Study 2: Process Intensification in Omalizumab Biosimilar (CMAB007)
The manufacturing optimization of the omalizumab biosimilar CMAB007 illustrates how medium- to high-risk upstream and downstream process improvements can be successfully validated using a science-based comparability approach. In this case, the manufacturer optimized the cell culture media and replaced the anion-exchange chromatography purification step, achieving an approximately fivefold increase in manufacturing productivity while retaining the original host cell line.
To support these manufacturing improvements, the sponsor implemented a tiered three-arm comparability strategy involving evaluation of pre-change material, post-change manufacturing batches, and fourteen commercial lots of the originator reference product (Xolair) obtained from several international markets. Comprehensive analytical characterization confirmed that every evaluated critical quality attribute remained within the predefined tier-specific acceptance ranges. This Chemistry, Manufacturing, and Controls (CMC) comparability package was further supported by a randomized, double-blind, two-arm pharmacokinetic bridging study involving 114 healthy male volunteers.
The clinical investigation demonstrated pharmacokinetic bioequivalence between the post-change biosimilar and Xolair. The geometric mean ratios (GMRs) and 90% confidence intervals (CI) for AUC₀–∞ (99.82%; 90% CI: 91.46%–108.94%), AUC₀–t (99.54%; 90% CI: 91.40%–108.41%), and Cmax (101.88%; 90% CI: 95.21%–109.01%) all remained comfortably within accepted regulatory bioequivalence criteria.
Comparative Global Standards and the Elimination of Clinical Efficacy Requirements
Regulatory authorities around the world have increasingly adopted the scientific position that comparative clinical efficacy studies should no longer serve as a routine requirement for biosimilar approval or post-approval manufacturing changes. This international consensus reflects the recognition that modern, high-resolution analytical characterization technologies possess significantly greater sensitivity for detecting manufacturing-related product differences than comparative clinical efficacy endpoints.
Several recent regulatory developments have accelerated this transition toward an analytical comparability-driven regulatory model.
United States FDA (October 2025 Updates)
The FDA’s updated guidance document, “Scientific Considerations in Demonstrating Biosimilarity to a Reference Product,” emphasizes that comparative clinical efficacy trials—which typically extend development timelines by approximately one to three years and add nearly 24 million to overall development costs—generally provide lower sensitivity than comprehensive analytical comparability assessments. When extensive analytical characterization demonstrates a high degree of similarity, an appropriately designed human pharmacokinetic similarity study combined with an immunogenicity assessment is generally considered sufficient to support regulatory approval.
Health Canada (May 2026 Shift)
Health Canada’s finalized guidance, “Guidance on Information and Submission Requirements for Biosimilar Biologic Drugs,” formally removed the routine requirement for Phase III comparative clinical efficacy studies in biosimilar development. Under this updated framework, detailed structural and functional characterization serves as the principal evidence supporting biosimilarity, enabling extrapolation of all approved reference product indications without requiring additional clinical justification.
European Medicines Agency (EMA Reflection Paper)
The European Medicines Agency’s draft Reflection Paper proposes a simplified regulatory pathway that allows comparative clinical efficacy studies to be waived when comprehensive analytical comparability, robust in vitro pharmacology data, and well-designed clinical pharmacokinetic studies collectively demonstrate biosimilarity. This science-driven approach is specifically intended for well-characterized biological proteins with clearly established mechanisms of action.
Read our case study on insulin biosimilar characterization to see our methodology in action.
Conclusion
The successful implementation of Post-Approval Changes for Approved Biosimilars relies on the integration of risk-based Quality-by-Design (QbD) principles, highly sensitive analytical characterization technologies, and robust control strategies developed in accordance with the ICH Q5E guideline. Together, these scientific approaches provide the analytical evidence necessary to support manufacturing improvements while maintaining confidence in product quality, safety, and clinical performance.
By collaborating with specialized analytical laboratories such as ResolveMass Laboratories Inc., biopharmaceutical manufacturers can develop phase-appropriate, orthogonally validated comparability programs that generate comprehensive structural characterization data suitable for regulatory review. These scientifically rigorous analytical packages help satisfy regulatory expectations, facilitate manufacturing process optimization, and support a consistent, reliable, and cost-effective supply of critical biological therapies.
For expert scientific consulting and advanced analytical testing services to support your post-approval manufacturing changes, contact the team at ResolveMass Laboratories Inc. by visiting the ResolveMass Contact Us page.
Frequently Asked Questions
Post-approval changes for approved biosimilars are generally categorized as major, moderate, or minor based on their potential effect on product quality, safety, purity, and potency. Major changes typically require a Prior Approval Supplement (PAS) before implementation, while moderate changes are submitted through a Changes Being Effected in 30 Days (CBE-30) notification. Minor modifications with minimal regulatory impact are usually documented in the Annual Report. This risk-based classification ensures that regulatory oversight is proportional to the significance of the manufacturing change.
Advanced analytical technologies can detect extremely small structural and functional changes in biological products that may not be observable through clinical efficacy studies. Techniques such as mass spectrometry, chromatography, and biophysical characterization provide highly sensitive molecular-level information, whereas clinical outcomes are often influenced by natural patient variability. Because of this greater sensitivity, regulatory agencies increasingly rely on analytical comparability data to demonstrate that manufacturing changes have not affected product performance.
A three-arm comparability strategy is particularly valuable for manufacturing changes that carry a higher scientific or regulatory risk, including host cell line replacements or major process modifications. This approach compares the post-change biosimilar with both the pre-change product and the original reference biologic to identify any cumulative product drift. By evaluating all three datasets together, manufacturers can demonstrate that the biosimilar continues to remain within the established quality profile throughout its lifecycle.
Yes. A host cell line transition may be approved without repeating comparative clinical efficacy studies when comprehensive analytical evidence demonstrates that the post-change product remains highly comparable to the original version. This evidence typically includes structural, physicochemical, functional, and biological characterization supported by pharmacokinetic (PK) bridging studies where appropriate. Regulatory authorities accept this science-based approach when the overall comparability package adequately addresses product quality and clinical performance.
A well-qualified in-house reference material serves as a consistent benchmark for monitoring product quality throughout the manufacturing lifecycle. It enables manufacturers to compare pre-change and post-change batches using a stable internal standard rather than relying on continuous testing against the originator reference product. This approach improves consistency in analytical evaluations while supporting long-term process control and regulatory compliance during routine manufacturing updates.
Health Canada manages post-NOC changes for biosimilars under the Food and Drug Regulations, where manufacturing modifications are generally submitted as either Supplements to a New Drug Submission (SNDS) or Notifiable Changes, depending on their level of risk. Following the updated guidance issued in May 2026, the agency emphasizes a quality-focused, science-based evaluation process that prioritizes comprehensive analytical comparability over routine comparative clinical efficacy studies whenever appropriate.
Higher-order structure is assessed using multiple complementary analytical techniques to confirm that protein folding and conformational stability remain unchanged after manufacturing modifications. Commonly used methods include Far- and Near-UV Circular Dichroism (CD), Differential Scanning Calorimetry (DSC), Fourier-Transform Infrared (FTIR) spectroscopy, and high-resolution 1D/2D Nuclear Magnetic Resonance (NMR) spectroscopy. Together, these orthogonal techniques provide detailed information on secondary structure, tertiary conformation, and thermal stability (Tm).
The FDA evaluates post-approval modifications for interchangeable biosimilars using the same risk-based comparability principles that apply to all licensed biological products. Manufacturing changes must be supported by appropriate analytical evidence demonstrating that product quality, safety, and potency remain unaffected. According to the FDA’s recent regulatory guidance, the scientific framework described in ICH Q5E is applicable to interchangeable biosimilars as well as standard biosimilars, ensuring a consistent regulatory approach.
Tier 1 quality attributes are assessed using formal statistical equivalence testing because these attributes have the greatest potential to influence clinical performance. The analysis determines whether the mean value of the post-change product remains within predefined equivalence limits, which are commonly established at ±1.5 standard deviations relative to the historical pre-change or reference product data. This statistical methodology is frequently applied to critical functional attributes such as biological activity and receptor-binding potency.
Introducing a biosimilar into a facility that manufactures multiple biological products requires a comprehensive risk assessment covering cross-contamination, equipment sharing, material movement, cleaning procedures, and personnel workflow. Manufacturers must establish validated cleaning protocols, product-specific identification methods, and effective contamination control strategies to maintain product integrity. Thorough facility risk assessments and ongoing process monitoring are essential to ensure compliance with current Good Manufacturing Practices (cGMP) and regulatory expectations.
Reference:
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use. (2004). Comparability of biotechnological/biological products subject to changes in their manufacturing process (ICH Harmonised Tripartite Guideline Q5E). https://database.ich.org/sites/default/files/Q5E%20Guideline.pdf
- European Medicines Agency. (2005). ICH Q5E: Comparability of biotechnological/biological products subject to changes in their manufacturing process – Step 5 (CPMP/ICH/5721/03). https://www.ema.europa.eu/en/documents/scientific-guideline/ich-q-5-e-comparability-biotechnologicalbiological-products-step-5_en.pdf
- Chow, S.-C., Song, F., & Bai, H. (2016). Analytical similarity assessment in biosimilar studies. The AAPS Journal, 18(3), 670–677. https://doi.org/10.1208/s12248-016-9882-5
- National Institutes of Health, SEED Innovator Support Team. (2024). Regulatory knowledge guide for biological products. https://seed.nih.gov/sites/default/files/2024-03/Regulatory-Knowledge-Guide-Biological-Products.pdf
- U.S. Food and Drug Administration. (2025, October). Scientific considerations in demonstrating biosimilarity to a reference product: Updated recommendations for assessing the need for comparative efficacy studies: Draft guidance for industry. https://www.fda.gov/media/180206/download
- Health Canada. (2026, May 19). Post-market information requirements: Guidance on information and submission requirements for biosimilar biologic drugs. Government of Canada. https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/applications-submissions/guidance-documents/information-submission-requirements-biosimilar-biologic-drugs/post-market.html
- Health Canada. (2019, December 19). Post-notice of compliance (NOC) changes: Quality guidance. Government of Canada. https://www.canada.ca/en/health-canada/services/drugs-health-products/drug-products/applications-submissions/guidance-documents/post-notice-compliance-changes/quality-document/guidance.html

