
Understanding Why Biosimilars Fail Regulatory Approval: An Analytical Overview
Biosimilars fail regulatory approval primarily because of deficiencies in chemistry, manufacturing, and controls (CMC), analytical comparability shortcomings, and manufacturing facility compliance issues rather than failures related to clinical efficacy. Successfully addressing these scientific and operational challenges is one of the most important determinants of achieving first-cycle approval and timely market entry.
Unlike small-molecule generic drugs, which are manufactured through defined chemical synthesis pathways that can reproduce identical active ingredients, biological therapeutics are produced within living host cell systems. As a result, exact replication of the reference biologic is not achievable. Regulatory authorities therefore require developers to demonstrate that a proposed biosimilar is “highly similar” to the reference product, with no clinically meaningful differences in safety, purity, or potency.
To understand the foundational requirements for these programs, learn more about our comprehensive biosimilar characterization services.
Historically, evaluating the underlying reasons for regulatory rejections was difficult because regulatory agencies did not publicly disclose Complete Response Letters (CRLs). This changed significantly in July 2025, when the U.S. Food and Drug Administration (FDA) introduced a public database containing more than 200 previously confidential CRLs. The released information revealed that biological product applications experience a disproportionately high rate of first-cycle non-approvals, with nearly half of all Biologics License Applications (BLAs) receiving a CRL before approval. Regulatory scrutiny has continued to intensify, with openFDA data indicating that 49 CRLs were issued for BLAs between 2023 and 2025, compared with only 22 during the 2020–2022 period.
For a deeper dive into the metrics that define success, explore our insights on biosimilar comparability studies.
This increase in regulatory oversight coincides with a major market transition. According to the IQVIA Institute, a significant “biosimilar void” is emerging between 2025 and 2034, during which 118 originator biologics representing approximately 234 billion in annual sales are expected to lose patent protection. However, by mid-2024, biosimilars were actively being developed for only 12 of those 118 molecules. The combination of high development failure rates and development costs ranging from 100 million to 300 million per biosimilar program has discouraged investment across the sector. For sponsors, analytical scientists, and regulatory professionals, understanding the technical causes of failure in areas such as CMC, glycosylation, charge heterogeneity, and facility compliance is essential for navigating this increasingly competitive and highly regulated environment.
Share via:
Article Summary:
- Biosimilar approvals are most often delayed or rejected because of chemistry, manufacturing, and controls (CMC) deficiencies, analytical comparability issues, and manufacturing compliance problems rather than lack of clinical effectiveness.
- Since biologics are produced in living cells, they cannot be copied exactly like traditional generic drugs. Regulators therefore require extensive evidence showing that a biosimilar is highly similar to the reference product in safety, quality, and performance.
- Common development pitfalls include structural differences from the reference product, inadequate analytical testing, protein aggregation, residual impurities, and formulation instability, all of which can raise concerns about safety, efficacy, or product consistency.
- Advanced analytical technologies, particularly high-resolution mass spectrometry and LC-MS/MS, are essential for detecting subtle molecular variations such as glycosylation changes, sequence variants, and post-translational modifications that could affect regulatory acceptance.
- Differences in glycosylation patterns and charge variants can significantly impact biological activity, pharmacokinetics, stability, and immunogenicity. Developers must demonstrate that these variations do not create clinically meaningful differences from the originator product.
- Manufacturing facility inspections play a major role in approval outcomes. GMP deficiencies, inadequate process validation, and quality system gaps can trigger regulatory rejections even when analytical and clinical data are otherwise acceptable.
- As regulators increasingly adopt analytical-first approval pathways, successful biosimilar developers must focus on robust process characterization, comprehensive comparability studies, strong GMP readiness, and early engagement with regulatory agencies to improve the likelihood of first-cycle approval.
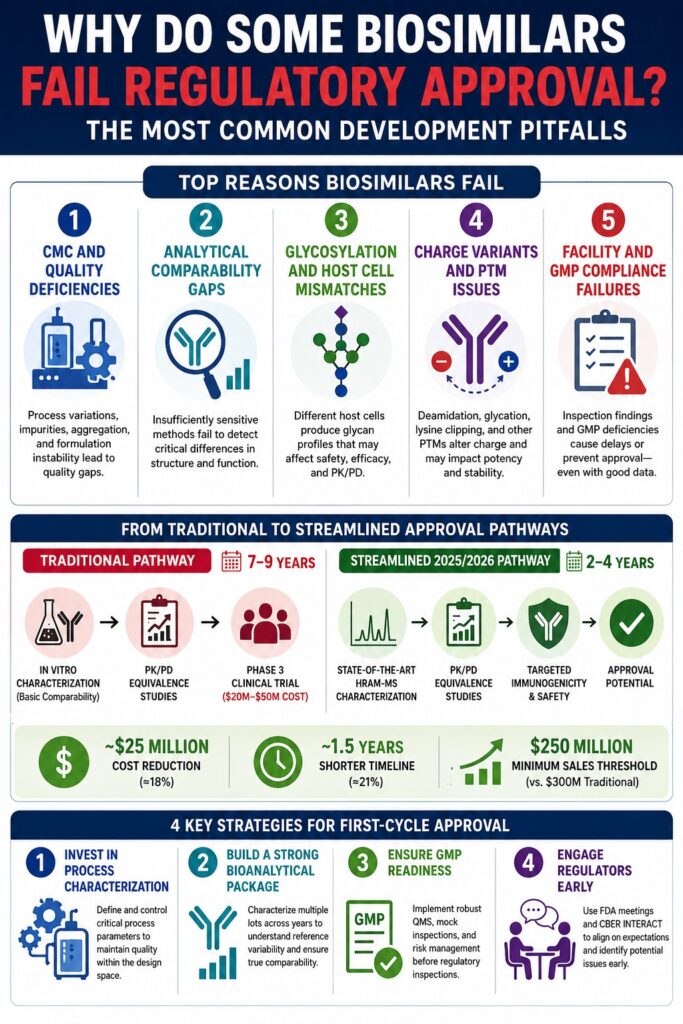
Primary CMC and Quality Pitfalls: Why Biosimilars Fail Regulatory Approval
Chemistry, manufacturing, and controls (CMC) deficiencies arise when upstream production processes or downstream purification operations fail to maintain structural and functional similarity to the reference product. These issues commonly occur because biological expression systems are extremely sensitive to even minor changes in manufacturing conditions and formulation strategies.
Biologics are highly complex glycoproteins, often exceeding 150 kD in molecular weight, whose biological function depends on intricate three-dimensional structures and sophisticated post-translational modifications (PTMs). Because the originator manufacturer’s cell line, media composition, process parameters, and purification methods are protected as proprietary information, biosimilar developers must effectively reverse-engineer the manufacturing process from the ground up.
Identify the potential challenges in your own pipeline by reviewing our guide on critical quality attributes (CQAs) in biosimilars.
Even subtle modifications to culture media, dissolved oxygen levels, nutrient feed strategies, or purification resins can influence critical quality attributes (CQAs). When these changes result in product characteristics that fall outside the established variability range of the reference product, regulatory agencies may issue a Complete Response Letter.
Common CMC-Related Failure Modes and Their Biological Impact
| Deficiency Category | Specific Molecular and Process Failures | Impact on Biological Performance | Regulatory Consequences |
|---|---|---|---|
| Comparability Failures | Structural differences in primary sequence, higher-order structure, or glycan distribution between biosimilar and reference lots | Alters receptor-binding kinetics, antibody-dependent cellular cytotoxicity (ADCC), or serum half-life | Can result in program suspension or extensive additional analytical characterization requirements |
| Analytical Method Gaps | Use of unvalidated, low-sensitivity, or single-attribute assays; inadequate orthogonal testing strategies | Prevents identification of low-abundance impurities, host cell proteins (HCPs), or structural variants | Refusal to File (RTF) decisions or CRLs due to incomplete analytical validation |
| Aggregation and Particulates | Elevated dimer or polymer formation levels greater than 1–2% resulting from processing or shipping stress | Increases risk of anti-drug antibody (ADA) generation and immunogenic responses | May trigger clinical holds or application rejection due to safety concerns |
| Impurity Profile Gaps | Inadequate removal of host cell proteins, host cell DNA, or extractables and leachables | May cause toxicity or inflammatory immune responses in patients | Requires extensive corrective and preventive action (CAPA) measures and process redesign |
| Formulation Instability | Poor excipient selection, pH instability, or vulnerability to environmental stress | Accelerates degradation, chemical modification, and protein unfolding | Can lead to reduced shelf-life approvals or outright rejection |
The Crucial Role of Mass Spectrometry in Preventing Regulatory Failure
Mass spectrometry plays a central role in preventing regulatory failure by enabling atomic-level characterization of biosimilars and confirming structural similarity to the reference biologic. High-resolution accurate mass (HRAM) platforms can detect deamidation, glycosylation changes, sequence variants, and structural allotypes that may remain undetected using conventional analytical approaches.
See how high-resolution data empowers your submission by exploring our intact mass analysis for biosimilars.
Analytical characterization serves as the foundation of the biosimilar approval pathway. If analytical methods lack sufficient sensitivity to resolve molecular micro-heterogeneity, the integrity of the entire regulatory submission can be compromised. To overcome these challenges, advanced bioanalytical laboratories increasingly rely on liquid chromatography-tandem mass spectrometry (LC-MS/MS) workflows to provide comprehensive structural characterization and minimize analytical uncertainty.
Discover how to enhance your precision by reviewing peptide mapping in biosimilars.
The Complexities of Glycosylation and Host Cell Mismatches
Glycosylation mismatches occur when the selected host cell line or manufacturing conditions generate glycan profiles that differ from those of the reference product. These differences can directly affect pharmacokinetic and pharmacodynamic performance.
During biomanufacturing, glycoproteins undergo highly regulated enzymatic modifications involving the attachment of carbohydrate chains at specific amino acid motifs, such as Asn-297 within the Fc region of IgG antibodies. Because these glycan structures influence biological activity, even minor changes can affect clinical performance.
A well-known example involves the reference product Remicade (infliximab), which is produced using a mouse myeloma Sp2/0 cell line. This expression system naturally generates terminal α-galactosylated and sialylated glycans, including N-glycolylneuraminic acid (NGNA). Many biosimilar developers instead utilize Chinese Hamster Ovary (CHO) cells because of their favorable productivity and scalability. However, CHO cells produce a different glycosylation profile and do not generate identical sialylated glycan species, resulting in an inherent glycan mismatch.
To satisfy regulatory requirements, developers must demonstrate that these glycosylation differences do not adversely affect safety, efficacy, or biological activity. This generally requires a combination of sophisticated analytical and functional techniques, including:
HILIC-FLD
Hydrophilic interaction liquid chromatography with fluorescence detection (HILIC-FLD) enables precise quantitative analysis of released glycan species and provides detailed glycan profiling data.
Reversed-Phase LC-MS/MS
Site-specific glycopeptide mapping using reversed-phase LC-MS/MS allows the identification and assignment of complex branched or isomeric glycan structures to precise amino acid locations within the protein sequence.
Functional Assays
Surface plasmon resonance (SPR) assays and cell-based bioassays help confirm that changes in core fucosylation or terminal galactosylation do not negatively affect FcγRIIIa receptor binding or ADCC activity.
Address these risks early by learning about our glycosylation analysis of biosimilars.
Charge Heterogeneity and Chemical Post-Translational Modifications
Uncontrolled charge heterogeneity is a common cause of regulatory rejection because chemical modifications occurring within biologically important regions of a therapeutic protein can substantially influence potency, stability, and pharmacological activity.
Charge variants arise through both enzymatic and non-enzymatic reactions during upstream production and downstream processing.
Acidic variants, characterized by lower isoelectric points (pI) and earlier elution during cation-exchange chromatography (CEX), commonly result from deamidation of asparagine residues to aspartic acid or isoaspartic acid, glycation, or terminal sialylation. When deamidation occurs within complementarity-determining regions (CDRs), antigen binding may be compromised, resulting in reduced therapeutic efficacy.
Basic variants, which display higher pI values, are often associated with incomplete C-terminal lysine clipping, succinimide formation, or amino acid substitutions. In one rituximab biosimilar characterization study, a candidate designated BC2 was ultimately rejected because it exhibited an amino acid substitution, significant alterations in N-glycosylation, and notable charge variant differences compared with the reference product.
Traditional charge variant analysis (CVA) relies heavily on liquid chromatography with ultraviolet detection (LC-UV). While effective for separating charge variants, LC-UV cannot identify the underlying molecular modifications responsible for those differences.
Modern analytical laboratories increasingly employ native mass spectrometry workflows using volatile buffer systems such as ammonium acetate or ammonium formate. These approaches allow direct coupling of CEX or capillary zone electrophoresis (CZE) with high-resolution Orbitrap mass spectrometers, enabling simultaneous separation, intact mass determination, and PTM identification within a single analytical workflow.
Traditional UV-Based CVA vs. Native MS-Based CVA
| Characterization Parameter | Traditional UV-Based CVA | Native MS-Based CVA (CEX-MS, CZE-MS) |
| Primary Identification Method | Indirect identification based on retention time comparisons | Direct identification using high-resolution accurate mass measurements |
| Separation Buffers | Non-volatile high-salt buffers that suppress ionization | Volatile MS-compatible buffers such as ammonium acetate and ammonium formate |
| Workflow Efficiency | Requires fraction collection and offline analysis | Automated online separation and identification in a single injection |
| Analytical Sensitivity | Limited ability to detect low-abundance variants below 1–5% | Detects micro-heterogeneous species down to approximately 0.1% abundance |
| Structural Insights | Minimal information regarding underlying modifications | Comprehensive identification of deamidation, glycation, oxidation, C-terminal lysine clipping, and disulfide pairing |
Upgrade your analytical capabilities with our specialized charge variant analysis for biosimilars.
Facility Inspection Gaps and the Pre-Approval Inspection Gauntlet
Pre-approval inspection (PAI) deficiencies and Good Manufacturing Practice (GMP) compliance failures frequently delay or prevent regulatory approval because agencies will not approve a product manufactured in facilities with unresolved quality concerns.
Industry data indicate that facility-related compliance issues contribute to more than 56% of all biologics and biosimilars Complete Response Letters. Notably, these rejections can occur even when analytical and clinical data packages are otherwise acceptable.
Achieving inspection readiness requires a quality-focused operational strategy that begins years before submission of the final Biologics License Application.
Recent Examples of Facility-Related Regulatory Challenges
Alvotech (AVT02, AVT03, AVT05)
Alvotech’s biosimilars referencing Humira (adalimumab), Prolia/Xgeva (denosumab), and Simponi (golimumab) experienced multiple regulatory setbacks because of unresolved observations identified during FDA inspections of the company’s Reykjavik manufacturing facility. FDA correspondence specifically noted that the deficiencies were unrelated to analytical similarity, safety, or efficacy.
Xbrane Biopharma (Lucant/Lucamzi)
In October 2025, Xbrane received a CRL concerning its ranibizumab biosimilar BLA referencing Lucentis. The rejection occurred despite demonstrated clinical equivalence and existing European Medicines Agency (EMA) approval. The FDA’s decision was based exclusively on unresolved manufacturing facility observations.
Biocon Biologics (Avastin Biosimilar)
Biocon Biologics and its partner Viatris received a CRL for their bevacizumab biosimilar referencing Avastin due to deficiencies identified during a manufacturing facility inspection.
These examples underscore an important industry reality: robust analytical and clinical data alone do not guarantee approval. Manufacturing readiness, technology transfer execution, and facility compliance are equally important determinants of regulatory success.
Even seemingly minor process modifications during scale-up, such as replacing a purification filter or transitioning from pilot-scale equipment to commercial-scale bioreactors, can affect shear stress, mixing efficiency, and pH distribution. Without comprehensive process validation and risk-based technology transfer strategies, such changes may lead to batch inconsistencies, sterility concerns, and regulatory rejection.
Ensure your processes meet global standards by consulting our experts on impurity profiling of biosimilars.
Navigating Shifting Regulatory Frameworks to Prevent Program Rejection
Sponsors can significantly reduce regulatory risk by aligning development programs with evolving global regulatory frameworks that increasingly prioritize analytical evidence over large-scale clinical efficacy trials.
Historically, biosimilar development followed a stepwise framework involving extensive analytical characterization, pharmacokinetic (PK) studies, and large comparative Phase 3 efficacy trials. Over time, however, regulatory agencies have concluded that comparative efficacy studies often contribute limited additional evidence when robust analytical and PK data are already available.
This recognition has resulted in major regulatory reforms.
FDA October 2025 Draft Guidance
The FDA released draft guidance titled Scientific Considerations in Demonstrating Biosimilarity to a Reference Product: Updated Recommendations for Assessing the Need for Comparative Efficacy Studies. Under this framework, biosimilars demonstrating strong analytical similarity through comparative analytical assessments (CAA), supported by human PK and immunogenicity data, may no longer require Phase 3 comparative efficacy studies.
Removal of Switching Studies
The October 2025 framework also eliminated the recommendation for complex switching studies previously required for interchangeable biosimilar designation, bringing biosimilar development closer to the generic drug approval model.
FDA March 2026 Revision 4 Q&As
In updated guidance from the Center for Drug Evaluation and Research (CDER), the FDA removed the default expectation that every biosimilar development program include direct clinical PK comparisons against a U.S.-licensed reference product. Under scientifically justified circumstances, developers may rely on data generated using non-U.S. comparators without conducting expensive three-way bridging studies.
EMA Tailored Clinical Approach (March 2026)
The EMA’s Committee for Medicinal Products for Human Use (CHMP) formally adopted a tailored clinical development approach establishing that comparative efficacy studies are generally no longer expected for most biosimilars when state-of-the-art analytical characterization adequately demonstrates biosimilarity.
Traditional Pathway vs. Streamlined Pathway
Traditional Development Pathway (7–9 Years)
- In Vitro Characterization (Basic Comparability)
- PK/PD Equivalence Studies
- Phase 3 Clinical Trial (20M–50M Cost)
Streamlined 2025/2026 Pathway (2–4 Years)
- State-of-the-Art HRAM-MS Characterization
- PK/PD Equivalence Studies
- Targeted Immunogenicity and Comparative Safety Assessment
The economic implications of these regulatory reforms are substantial. Financial analyses of monoclonal antibody biosimilar programs suggest that Phase 3 trial waivers can reduce development expenses by approximately 25 million per program, representing an 18% cost reduction. Development timelines may be shortened by approximately 1.5 years, equating to a 21% reduction in overall program duration.
Additionally, the minimum peak sales threshold required for commercial viability may decline from 300 million to 250 million, expanding opportunities for biosimilars targeting niche and orphan disease indications.
However, these streamlined pathways are contingent upon exceptional analytical evidence. Sponsors cannot rely on clinical studies to compensate for analytical deficiencies. Any residual uncertainty regarding molecular similarity or biological activity may still prompt regulators to require additional clinical evaluation.
Consequently, development strategies must increasingly prioritize advanced analytical characterization and high-resolution comparability assessments.
Strategic Recommendations for Biosimilar Developers: The Path to First-Cycle Approval
The most reliable route to first-cycle approval involves comprehensive characterization of the reference product’s historical variability combined with proactive regulatory engagement throughout development.
Adherence to Quality by Design (QbD) principles helps ensure that structural similarity is incorporated into the manufacturing process from the earliest stages of development.
1. Invest in Comprehensive Process Characterization
Developers should implement robust upstream and downstream process characterization programs. High-throughput screening and QbD methodologies help define process design spaces where critical process parameters (CPPs), including temperature, pH, dissolved oxygen, and nutrient feed rates, remain tightly controlled.
Maintaining consistency at the process level is essential for preserving stable glycosylation profiles and charge variant distributions.
2. Build a Comprehensive Bioanalytical Characterization Package
Because biosimilarity is assessed against a range of reference product variability rather than a single target value, developers should obtain and characterize numerous reference product lots collected across multiple years.
This extensive dataset helps define the originator’s variability profile and reduces the risk of targeting a non-representative quality range that could result in comparability deficiencies.
3. Prioritize cGMP Readiness and Technology Transfer Validation
All manufacturing facilities, including contract development and manufacturing organizations (CDMOs), should establish GMP readiness early in development.
Routine mock inspections, comprehensive Quality Management Systems (QMS), Failure Mode and Effects Analysis (FMEA), and proactive risk mitigation programs can identify vulnerabilities before official inspections occur.
4. Maintain Active Regulatory Engagement
Developers should take advantage of FDA Type B, Type C, and Type D meetings, as well as programs such as CBER INTERACT, to obtain product-specific guidance regarding analytical expectations, CMC readiness, and opportunities to qualify for clinical study waivers.
Regular interaction with regulatory authorities helps identify potential deficiencies early and ensures alignment with evolving regulatory expectations.
Nuanced Conclusions: Overcoming the Factors Behind Biosimilar Regulatory Failure
The central lesson regarding why biosimilars fail regulatory approval is that successful clinical outcomes ultimately depend on rigorous molecular characterization, robust quality control systems, and strong CMC execution. Sponsors that prioritize analytical characterization and formulation stability from the outset are better positioned to leverage modern regulatory pathways, reduce development risk, and accelerate commercialization.
As regulatory agencies such as the FDA and EMA continue moving toward analytical-first approval models, the demand for sophisticated, high-resolution analytical data has never been greater. Biosimilar developers increasingly require specialized bioanalytical partners capable of generating comprehensive, regulatory-compliant datasets that support streamlined approval pathways.
ResolveMass Laboratories Inc. serves as a leading contract research partner uniquely positioned to help biosimilar developers reduce risk and achieve first-cycle approvals. Through comprehensive pre-formulation studies, formulation development and optimization, and scale-up technology transfer support, the company helps ensure that drug products are designed for stability, quality, and therapeutic equivalence from the earliest stages of development.
ResolveMass Laboratories Inc. also specializes in reverse-engineering reference listed drugs (RLDs), designing robust formulations, and performing advanced excipient compatibility studies using data-driven Quality by Design methodologies.
Most importantly, ResolveMass Laboratories Inc. provides advanced charge variant analysis along with intact, subunit, and peptide-level mass spectrometry characterization. By leveraging native-MS workflows and state-of-the-art Orbitrap instrumentation, the laboratory delivers highly sensitive and validated analytical comparability data that supports clinical trial waivers, facilitates compliance with evolving global regulatory frameworks, and helps developers avoid the common pitfalls that lead to biosimilar regulatory failure.
To accelerate development timelines, strengthen CMC compliance, and collaborate with experienced bioanalytical specialists, visit the ResolveMass Laboratories Inc. Contact Us page to discuss your formulation development and analytical characterization requirements.
Frequently Asked Questions
Most first-cycle biosimilar rejections are linked to deficiencies in Chemistry, Manufacturing, and Controls (CMC), inadequate analytical comparability data, or manufacturing facility compliance issues rather than problems with clinical effectiveness. Since biologics are produced in living cells, even small changes in manufacturing conditions can alter critical quality attributes. Regulatory agencies closely evaluate molecular similarity, including glycosylation patterns and charge variants. When these attributes fall outside the accepted range of the reference product, additional data or corrective actions are usually required before approval can be granted.
A Complete Response Letter (CRL) is an official FDA communication stating that an application cannot be approved in its current form after review has been completed. The letter identifies specific deficiencies related to areas such as manufacturing processes, facility inspections, analytical validation, or clinical evidence. Developers must address all listed concerns before resubmitting the application. Receiving a CRL can significantly delay product launch timelines and increase development expenses due to additional studies, remediation efforts, and regulatory review cycles.
The choice of host cell line plays a critical role in determining the final structure and quality of a biologic product. Different cell lines can generate distinct post-translational modifications, glycosylation profiles, and impurity patterns. When a biosimilar is produced using a different expression system than the reference product, analytical differences may emerge that require detailed evaluation. Developers must demonstrate through extensive characterization and functional testing that these variations do not affect safety, efficacy, or pharmacokinetic performance.
Recent FDA guidance reflects a growing emphasis on advanced analytical characterization rather than routine Phase 3 comparative efficacy studies. Under the updated framework, developers may qualify for a waiver if they can demonstrate strong analytical similarity, human pharmacokinetic equivalence, and acceptable immunogenicity outcomes. This approach recognizes that highly sensitive analytical methods often provide more meaningful evidence of biosimilarity than large clinical efficacy trials. As a result, development timelines and overall program costs can be substantially reduced.
Pre-approval inspections remain one of the most common reasons for delayed biosimilar approvals. Even when analytical, nonclinical, and clinical data satisfy regulatory expectations, unresolved cGMP observations at manufacturing facilities can prevent approval. Several high-profile biosimilar programs have received Complete Response Letters because of inspection findings rather than product-related concerns. These cases highlight the importance of maintaining inspection readiness and strong quality systems throughout the development process.
Clinical efficacy studies are designed to assess outcomes in diverse patient populations and typically use broad equivalence margins. Because of this, they may not detect subtle structural differences between a biosimilar and its reference product. High-resolution mass spectrometry, on the other hand, can identify minute variations in molecular structure, post-translational modifications, and impurity profiles at extremely low levels. This analytical precision makes mass spectrometry one of the most powerful tools for demonstrating biosimilarity and supporting regulatory submissions.
Glycosylation is considered a critical quality attribute because it can directly influence a biologic’s function, stability, pharmacokinetics, and immune response profile. Even small changes in glycan composition may affect receptor interactions, antibody-dependent cellular cytotoxicity (ADCC), or drug clearance rates. Regulatory agencies therefore require detailed glycosylation characterization to confirm similarity with the reference product. Comprehensive glycan analysis helps ensure that the biosimilar performs consistently and maintains an acceptable safety profile.
Charge variant analysis (CVA) is used to evaluate acidic and basic forms of a therapeutic protein that arise during manufacturing and storage. These variants can result from modifications such as deamidation, glycation, or incomplete C-terminal lysine processing. Significant differences in charge distribution may affect protein stability, biological activity, or therapeutic performance. Demonstrating that the biosimilar exhibits a charge variant profile comparable to the reference product is therefore an important component of analytical similarity assessments.
Reference:
- U.S. Food and Drug Administration. (2025, October 29). FDA moves to accelerate biosimilar development and lower drug costs. U.S. Department of Health and Human Services. FDA moves to accelerate biosimilar development and lower drug costs
- Agbogbo, F. K., Ecker, D. M., Farrand, A., Han, K., Khoury, A., Martin, A., McCool, J., Rasche, U., Rau, T. D., Schmidt, D., Sha, M., & Treuheit, N. (2019). Current perspectives on biosimilars. Journal of Industrial Microbiology & Biotechnology, 46(9–10), 1297–1311. https://doi.org/10.1007/s10295-019-02216-z
- U.S. Food and Drug Administration. (2022, April). Considerations in demonstrating interchangeability with a reference product: Guidance for industry. U.S. Department of Health and Human Services. https://www.fda.gov/media/154919/download
- U.S. Food and Drug Administration. (2025, September 4). FDA announces real-time release of complete response letters, posts previously unpublished batch of 89. U.S. Department of Health and Human Services. https://www.fda.gov/news-events/press-announcements/fda-announces-real-time-release-complete-response-letters-posts-previously-unpublished-batch-89
- Rathore, A. S., & Malani, H. (2025). Predicting biosimilar comparability using glycosylation, charge variants, potency, and stability attributes. ResearchGate. https://www.researchgate.net/publication/405476342_Predicting_Biosimilar_Comparability_Using_Glycosylation_Charge_Variants_Potency_and_Stability_Attributes
- Mori, K., Ishii-Watabe, A., & Aoyama, M. (2025). Characterization of biosimilar monoclonal antibodies and their reference products approved in Japan to reveal the quality characteristics in post-approval phase. Biologicals, 98, 101756. https://doi.org/10.1016/j.biologicals.2025.101756
- U.S. Food and Drug Administration. (2025, October 29). Scientific considerations in demonstrating biosimilarity to a reference product: Updated recommendations for assessing the need for comparative efficacy studies (Draft guidance for industry). U.S. Department of Health and Human Services. https://www.fda.gov/media/189366/download

