
Introduction
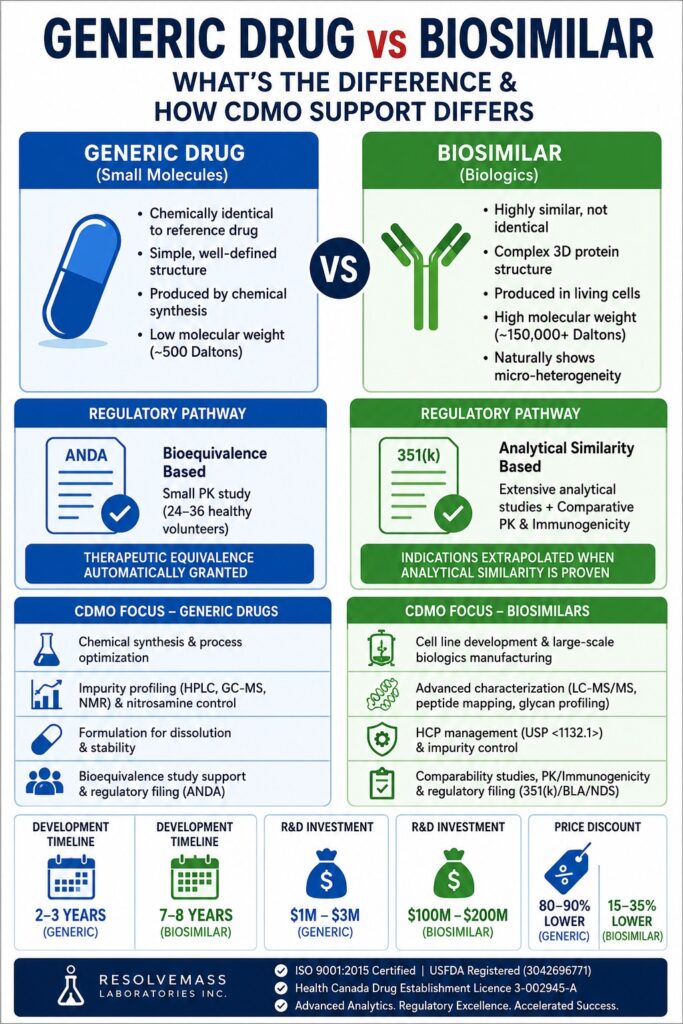
The distinction between a Generic Drug vs Biosimilar forms the core scientific and regulatory boundary separating chemically synthesized small-molecule medicines from highly complex biologic therapeutics. Generic drugs are chemically identical versions of their reference brand-name products, whereas biosimilars are highly similar therapeutic proteins produced in living cells and naturally exhibit micro-heterogeneity. Owing to these substantial differences in molecular origin and structural complexity, a therapeutic product can be classified as either a generic drug or a biosimilar—but never both.
For pharmaceutical manufacturers and biotechnology organizations worldwide, selecting the appropriate Contract Development and Manufacturing Organization (CDMO) depends on aligning these distinct molecular categories with specialized scientific expertise. Generic drug development relies on comprehensive solid-state characterization and well-designed bioequivalence studies, while biosimilar development demands sophisticated cell-line engineering, advanced purification technologies, and extensive structural comparability assessments. Therefore, an effective CDMO must possess the analytical precision, regulatory expertise, and state-of-the-art instrumentation necessary to successfully support these two fundamentally different categories of follow-on therapeutics.
Discover how our tailored development platforms can accelerate your molecule’s journey to market by exploring our comprehensive Biosimilar Characterization Services.
Article Summary:
- Generic drugs and biosimilars are fundamentally different therapeutic products. Generic medicines are chemically identical copies of small-molecule drugs, whereas biosimilars are highly similar versions of complex biologic medicines produced using living cells, making exact molecular replication impossible.
- Their development follows distinct scientific and manufacturing approaches. Generic drugs rely on chemical synthesis and bioequivalence testing, while biosimilars require advanced cell culture processes, protein purification, glycosylation analysis, and comprehensive structural characterization.
- Regulatory approval pathways are based on different evidence requirements. Generic products are primarily approved through pharmacokinetic bioequivalence studies, whereas biosimilars must demonstrate analytical, structural, and functional similarity using a totality-of-evidence approach, with extensive clinical efficacy trials often no longer required.
- Analytical testing for biosimilars is considerably more complex. In addition to conventional quality testing, biosimilar development requires advanced technologies such as LC-MS/MS, peptide mapping, glycan profiling, higher-order structure analysis, and host cell protein characterization to confirm product comparability.
- CDMO responsibilities differ according to the product type. Generic drug projects focus on formulation optimization, impurity profiling, stability testing, and regulatory submissions, while biosimilar programs demand expertise in biologics manufacturing, process development, analytical comparability, and regulatory compliance.
- Product stability and substitution policies also vary significantly. Generic drugs are generally easier to formulate and are widely eligible for automatic pharmacy substitution, whereas biosimilars require specialized formulations to preserve protein stability, and interchangeability depends on country-specific regulatory policies.
- Choosing the right CDMO is essential for successful product development. Organizations with advanced analytical capabilities, strong regulatory knowledge, and experience in both small-molecule and biologic characterization help pharmaceutical companies accelerate development while meeting evolving global quality and compliance standards.
Scientific Distinctions in the Generic Drug vs Biosimilar Paradigm
The scientific differences within the Generic Drug vs Biosimilar paradigm arise from the contrast between chemically synthesized small molecules and biologically expressed large-molecule therapeutics. Generic drugs are stable, low-molecular-weight compounds that can be comprehensively characterized using conventional analytical techniques. In contrast, biosimilars are large, intricately folded proteins produced by living cells and inherently display structural variability.
Molecular Weight and Structural Heterogeneity
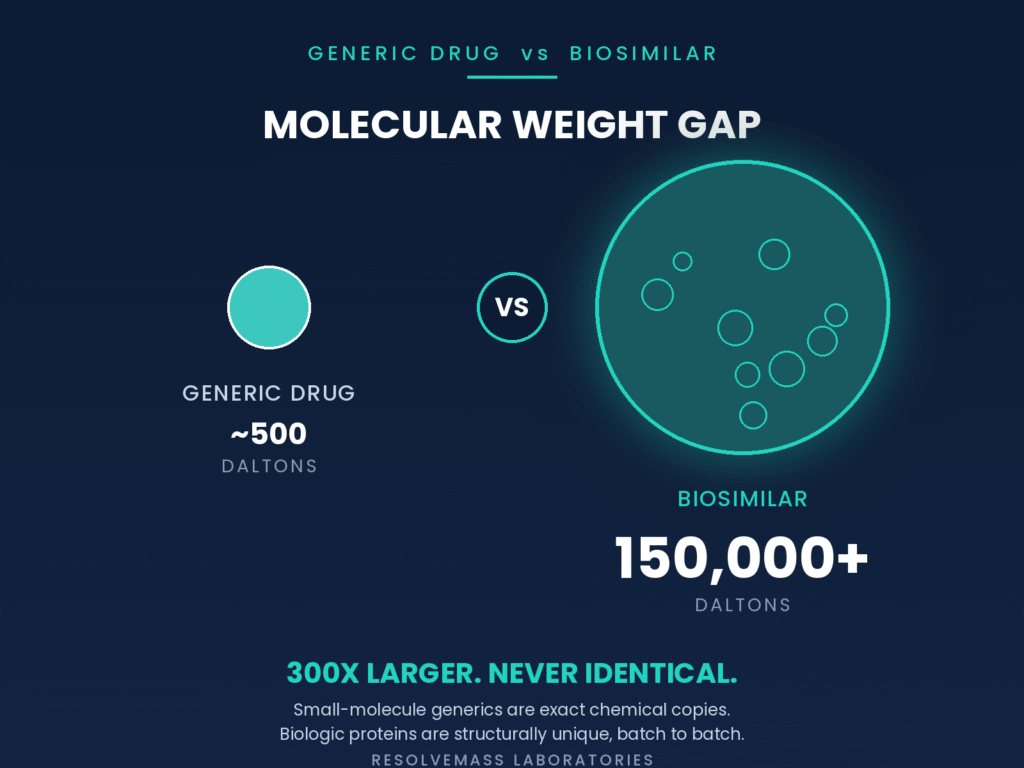
Molecular weight and structural heterogeneity serve as defining characteristics that distinguish these therapeutic classes. Generic small-molecule drugs are low-molecular-weight chemical entities that can be reproduced with identical composition, whereas biosimilars consist of large, three-dimensional proteins that naturally contain structural variations. Most generic drugs possess molecular weights of approximately 500 Daltons, while biologic proteins commonly exceed 150,000 Daltons. This dramatic difference in molecular size means that a small-molecule drug has a precisely defined chemical structure that can be duplicated exactly, whereas a biosimilar represents a highly sophisticated biological product that cannot be reproduced with absolute molecular identity.
Small Molecule (Generic) Biologic / Biosimilar (Complex Protein)
[~500 Daltons] [~150,000+ Daltons]
- Simple, well-defined structure – Highly folded three-dimensional conformation
- Produced through chemical synthesis – Produced through cellular expression
- Chemically identical copies – Heterogeneous pool of glycoforms
Because biologics are manufactured using living expression systems—including Chinese Hamster Ovary (CHO) cells, yeast, or bacterial hosts—the resulting proteins undergo an extensive range of post-translational modifications (PTMs). These modifications include N-glycosylation, C-terminal lysine clipping, deamidation, oxidation, and disulfide bond formation. Even batches of the same originator biologic naturally contain minor molecular variations, meaning the active pharmaceutical ingredient exists as a heterogeneous population of closely related glycoforms rather than perfectly identical molecules. Consequently, biosimilar developers must accurately reproduce the glycoform distribution and structural fingerprint of the Canadian Reference Biologic Drug (CRBD) or the corresponding U.S. reference biologic.
Optimize your early-stage upstream processes by reviewing our advanced workflows for Cell Line Development for Biosimilars.
Post-Translational Modifications and Glycoform Mapping
Post-translational modifications and glycoform mapping are exclusive considerations for biologic therapeutics because proteins produced by living cells are modified with complex carbohydrate structures that directly influence their biological function and clinical performance. These modifications present one of the greatest scientific challenges during biosimilar development, as variations in glycosylation patterns can significantly affect immunogenicity, serum half-life, receptor binding, and overall therapeutic activity.
Gain deep insights into how structural alterations impact safety and efficacy with our specialized guide on Post-Translational Modifications (PTMs) in Biosimilars.
To comprehensively evaluate these structural features, CDMOs employ advanced chromatographic and mass spectrometry-based analytical workflows. Hydrophilic Interaction Liquid Chromatography coupled with Fluorescence Detection (HILIC-FLD) provides highly accurate relative quantification of glycoform populations, while mass spectrometry (MS) enables direct structural identification of individual glycans. Employing these complementary analytical platforms is essential because glycosylation patterns can substantially influence ionization efficiency during MS analysis. As a result, orthogonal analytical approaches provide more reliable relative abundance measurements and improve confidence in glycoform characterization.
Ensure accurate carbohydrate profiling for your therapeutic protein with our targeted Glycosylation Analysis of Biosimilars.
Regulatory Approvals and Abbreviated Filing Strategies
Regulatory approval pathways differ significantly between generic drugs and biosimilars. Generic drug approvals primarily depend on demonstrating bioequivalence, whereas biosimilar approvals require extensive analytical comparability studies. These distinct regulatory frameworks reflect the fundamentally different scientific risks associated with chemically synthesized molecules versus biologically derived proteins.
Bioequivalence Pathways for Generic Drugs
Bioequivalence pathways for generic drugs require manufacturers to submit Abbreviated New Drug Applications (ANDAs) demonstrating that the proposed generic product delivers the same rate and extent of systemic exposure as the approved reference drug. Under the Hatch-Waxman Act in the United States and the Food and Drug Regulations in Canada, manufacturers can avoid repeating lengthy clinical efficacy trials by successfully establishing bioequivalence with an already-approved reference listed drug.
The central objective of generic bioequivalence studies is to confirm that the generic formulation delivers the same amount of Active Pharmaceutical Ingredient (API) into systemic circulation at an equivalent rate compared with the branded product. These studies are generally conducted in relatively small clinical trials involving 24 to 36 healthy volunteers. Therapeutic equivalence is established when pharmacokinetic parameters—including the Area Under the Curve (AUC) and the maximum plasma concentration (Cmax)—fall within the accepted regulatory acceptance interval of 80% to 125%. Once these requirements are met, the generic product is generally eligible to claim all approved therapeutic indications of the reference medicine.
Review the rigorous requirements for establishing therapeutic equivalence with our analysis of Comparative Testing Between Generic and RLD.
The Evolving Landscape of Biosimilar Comparability Exercises
The evolving regulatory framework for biosimilars places greater emphasis on analytical similarity than on large comparative clinical efficacy trials, consistent with updated guidance from Health Canada and the US FDA. Since biologics cannot be reproduced as exact molecular copies, biosimilar developers must provide a comprehensive evidence package following the totality-of-the-evidence approach to demonstrate that no clinically meaningful differences exist in safety, purity, or potency compared with the reference biologic.
The biosimilar approval pathway has undergone substantial modernization in recent years. Previously, both Health Canada and the US FDA routinely required comparative Phase III clinical efficacy and safety studies to establish biosimilarity. However, finalized regulatory guidance—including Health Canada’s May 19, 2026 update—indicates that these Phase III comparative clinical studies are generally unnecessary when the biosimilar has been thoroughly characterized and demonstrated to be highly similar using sensitive structural and functional analytical methodologies.
Learn how to navigate the complex regulatory expectations for demonstrating biosimilarity in our detailed overview of the Comparability Exercise in Biosimilar Development.
The US FDA and the European Medicines Agency (EMA) have adopted similar regulatory perspectives, recognizing that advanced analytical technologies possess greater sensitivity for detecting meaningful product differences than broad clinical outcome studies. Consequently, regulatory expectations now emphasize comparative pharmacokinetic evaluations in healthy participants, supported by comprehensive immunogenicity assessments.
| Regulatory Parameter | Generic Drugs (ANDA / ANDS) | Biosimilars (351(k) / NDS / BLA) |
|---|---|---|
| Primary Regulatory Focus | Chemical identity and pharmacokinetic bioequivalence | Structural and functional analytical similarity |
| Clinical Efficacy Trials | Not required | Typically waived under modern analytical guidance |
| Pharmacokinetic Study Scope | Generally 24–36 healthy volunteers | Comparative PK studies demonstrating equivalence |
| Extrapolation of Indications | Automatically applies to approved indications | Granted without additional justification when analytical similarity is established |
Strategic CDMO Support for Generic Drug vs Biosimilar Pipelines
CDMO support for a Generic Drug vs Biosimilar development program differs substantially depending on whether the product involves reverse engineering of chemically synthesized compounds or comprehensive characterization of recombinant biologic proteins. The required operational infrastructure, scientific expertise, manufacturing capabilities, and quality management systems vary considerably between these two development pathways.
Analytical Challenges in the Generic Drug vs Biosimilar Validation
Analytical challenges encountered during Generic Drug vs Biosimilar validation arise from the differences between relatively straightforward chromatographic evaluation of small molecules and comprehensive multidimensional characterization of complex biologics. For generic drugs, CDMO analytical activities primarily focus on confirming chemical identity, validating chromatographic methods, assessing purity, and profiling synthetic impurities to match the reference listed drug. In contrast, biosimilar development requires sophisticated analytical platforms, including LC-MS/MS, peptide mapping, glycan profiling, and functional bioassays to establish structural and functional comparability.
For small-molecule generic products, CDMOs routinely employ High-Performance Liquid Chromatography (HPLC), Gas Chromatography-Mass Spectrometry (GC-MS), and Nuclear Magnetic Resonance (NMR) spectroscopy to identify and quantify organic, inorganic, and volatile impurities. Particular attention is given to controlling mutagenic impurities such as nitrosamines, which requires optimized synthetic route design, kinetic process evaluation, and continuous monitoring throughout manufacturing.
In comparison, biopharmaceutical CDMOs must address complex process-related impurities, particularly Host Cell Proteins (HCPs). Conventional ELISA methods provide an estimate of overall HCP concentration but offer limited information regarding individual protein identities. According to the updated USP <1132.1> guidance, advanced mass spectrometry enables the identification and quantification of specific HCP species. This capability has become increasingly important because regulatory agencies now seek detailed information regarding exactly which HCPs remain within the drug substance, rather than only measuring the total residual HCP content.
Traditional Assay (ELISA) Advanced Mass Spectrometry (LC-MS/MS)
- Broad measurement of total HCP content – Direct identification of individual proteins
- Influenced by antibody coverage limitations – Detects specific high-risk HCPs
- Simple colorimetric measurement – Precise and absolute protein quantification
Streamline your contamination profiling strategy by checking out our specialized approach to Impurity Profiling of Biosimilars.
Advanced Analytical Characterization and Reverse Engineering
Advanced analytical characterization depends on high-resolution mass spectrometry and complementary spectroscopic techniques to establish the complete molecular fingerprint of a candidate product relative to its reference medicine. In biosimilar development, these characterization strategies include confirming both the primary amino acid sequence and higher-order structure (HOS) through technologies such as native MS, ion-mobility MS, and hydrogen-deuterium exchange mass spectrometry (HDX-MS).
Explore how to maintain native protein conformations during mass spectrometry with our breakdown of Native Mass Spectrometry for Biosimilars.
Peptide mapping continues to serve as the gold standard for verifying the primary protein sequence. This workflow begins with meticulous sample preparation, including denaturation, reduction, alkylation, and enzymatic digestion using highly specific proteases such as trypsin, LysC, GluC, or pepsin. The generated peptide fragments are subsequently separated using reversed-phase liquid chromatography (RPLC) with advanced bio-inert titanium-based column hardware designed to minimize non-specific interactions. Finally, the peptides are analyzed using high-resolution accurate mass (HRAM) mass spectrometers, including Quadrupole Time-of-Flight (Q-ToF) instruments, to verify sequence integrity and accurately identify the location of post-translational modifications.
Ensure 100% primary sequence coverage and precise modification site identification through our advanced protocol for Peptide Mapping in Biosimilars.
Host Cell Protein (HCP) Management and USP <1132.1>
Host cell protein (HCP) management is now guided by the updated USP <1132.1> chapter, which establishes high-resolution LC-MS/MS as an orthogonal analytical approach alongside conventional ELISA. Officially implemented on May 1, 2025, this regulatory chapter defines clear expectations for mass spectrometry-based method validation, system suitability testing, and quantitative analysis of residual HCPs.
The USP <1132.1> framework outlines three validated strategies for the quantification of residual host cell proteins:
Relative to Product Protein:
This approach uses peptides derived from the product protein at known concentrations as internal references. Residual HCP levels are estimated by comparing the signal intensity of HCP peptides with those of the product protein. Although this method is straightforward and efficient, quantitative accuracy may be influenced by differences in peptide ionization efficiency.
Relative to Spiked-In Proteins:
This strategy introduces intact reference proteins that do not interfere with the sample prior to enzymatic digestion. These external protein standards compensate for variability during sample preparation and digestion, providing a reliable semi-quantitative assessment of the overall HCP profile.
Relative to Spiked-In Peptides:
This methodology employs stable isotope-labeled synthetic peptides corresponding to targeted high-risk HCPs. It represents the highest level of analytical accuracy by enabling absolute, target-specific quantification of individual host cell proteins with exceptional precision.
The integration of these LC-MS/MS-based quantification approaches enables CDMOs to strengthen process development by identifying persistent “hitchhiker” proteins that remain associated with the therapeutic product throughout purification. This enhanced level of characterization improves process optimization, supports regulatory compliance, and contributes to maintaining patient safety.
See how high-resolution proteomics workflows can enhance your impurity detection threshold by reading about our Proteomics Approach for Biosimilars.
Formulation Chemistry and Stability Profiles
Formulation chemistry is primarily focused on maintaining the physical and chemical stability of the finished product, a task that is considerably more demanding for biologic proteins than for conventional small-molecule drugs. Generic formulations are generally designed to achieve dissolution characteristics equivalent to the reference product using established pharmaceutical excipients. In contrast, biosimilar formulations must continuously protect the highly sensitive three-dimensional structure of therapeutic proteins throughout manufacturing, storage, transportation, and administration.
| Formulation Attribute | Small-Molecule Generics | Large-Molecule Biosimilars |
|---|---|---|
| Formulation Objective | Rapid dissolution and equivalent release profile | Structural preservation and prevention of protein aggregation |
| Typical Excipients | Diluents, binders, disintegrants, and lubricants | Buffers, non-ionic surfactants, sugars, and amino acids |
| Primary Degradation Risks | Chemical hydrolysis, photolysis, and oxidation | Deamidation, oxidation, aggregation, and denaturation |
| Packaging Requirements | Conventional bottles or blister packaging for solid oral dosage forms | Prefilled syringes, vials, or autoinjectors |
Because therapeutic proteins are extremely susceptible to both physical and chemical stress, biopharmaceutical CDMOs must carefully optimize formulation compositions by selecting stabilizing excipients such as polysorbates and trehalose to reduce surface adsorption, aggregation, and conformational instability. Furthermore, comprehensive forced degradation studies are performed to compare degradation pathways between the biosimilar candidate and its reference biologic under a variety of stress conditions, including elevated temperatures, mechanical agitation, and repeated freeze-thaw cycles. These studies help confirm comparable stability profiles and support equivalent shelf-life performance.
Assess the long-term stability and degradation pathways of your biologic candidate using our structured protocols for Forced Degradation of Biosimilars.
Clinical Interchangeability and Substitution Frameworks
Clinical interchangeability determines whether pharmacists are permitted to substitute one therapeutic product for another without obtaining prior approval from the prescribing healthcare provider. While automatic substitution is well established for chemically synthesized generic drugs, substitution policies for biosimilars remain more complex and are governed by evolving regulatory frameworks.
Automatic Substitution for Generic Medications
Automatic substitution of generic medications is broadly authorized across most healthcare systems worldwide. Since the active pharmaceutical ingredient in a generic drug is chemically identical to that of the branded reference product, it is considered therapeutically equivalent and is expected to provide the same efficacy and safety profile. As a result, pharmacists routinely replace branded medicines with approved generic alternatives during dispensing, generating healthcare cost savings that commonly range from 80% to 90% for both patients and insurance providers.
Interchangeability and Pharmacy-Level Substitution for Biosimilars
For biosimilars, pharmacy-level substitution is generally not automatic unless the product has received a formal “interchangeable” designation from a regulatory authority such as the US FDA. Under the FDA’s 351(k) biosimilar pathway, manufacturers seeking this designation must demonstrate that repeated switching between the biosimilar and its reference biologic does not increase safety risks or reduce therapeutic effectiveness compared with continuous treatment using only the reference product.
By comparison, Health Canada and the European Medicines Agency (EMA) do not assign a separate interchangeability designation. Instead, these regulatory authorities recognize all approved biosimilars as highly similar and clinically equivalent to their respective reference biologics. Nevertheless, decisions regarding automatic pharmacy substitution remain the responsibility of individual provinces, territories, or member states, resulting in jurisdiction-specific substitution policies.
Examine our technical approach to establishing full clinical and analytical parity through our Biosimilar Comparability Studies.
Comparative Technical Specifications: Generic Drug vs Biosimilar
A comparison of technical development parameters highlights the substantial differences in timelines, financial investment, manufacturing complexity, and regulatory expectations between generic drugs and biosimilars. The relative simplicity of chemically synthesized small molecules allows for shorter development cycles, whereas biologic drug development requires extensive infrastructure, sophisticated manufacturing platforms, and significant financial investment.
| Parameter | Generic Drugs (Small Molecules) | Biosimilars (Biologics) |
|---|---|---|
| Development Timeline | Typically 2 to 3 years | Typically 7 to 8 years |
| Average R&D Investment | 1 million to 3 million | 100 million to 200 million |
| Primary Regulatory Filing | Abbreviated New Drug Application (ANDA) | Biologics License Application (BLA) / 351(k) / NDS |
| Active Ingredient Identity | Identical chemical duplicate | Highly similar product with natural batch-to-batch micro-heterogeneity |
| Clinical Development Scope | Small-scale PK/PD bioequivalence studies in healthy volunteers | PK/PD equivalence, immunogenicity assessment, with comparative clinical efficacy studies generally bypassed under modern regulatory pathways |
| Manufacturing Platform | Chemical synthesis using highly controlled reactors | Genetically engineered living cell cultures grown in bioreactors |
| Average Market Discount | 80% to 90% lower than the originator brand | 15% to 35% lower than the reference biologic |
Conclusion: Selecting an Analytical Partner for Generic Drug vs Biosimilar Pipelines
Successfully advancing Generic Drug vs Biosimilar development programs requires partnering with a CDMO that possesses the scientific expertise, advanced analytical capabilities, and regulatory qualifications appropriate for the specific molecular class under development. As global regulatory frameworks continue to evolve, analytical science has become the primary foundation for demonstrating product quality, similarity, and regulatory compliance. High-resolution mass spectrometry, comprehensive impurity characterization, and rigorously validated analytical methods now play a decisive role in obtaining regulatory approval.
To reduce development risks and accelerate product commercialization, pharmaceutical sponsors should collaborate with a CDMO that combines extensive scientific experience with proven regulatory compliance. ResolveMass Laboratories Inc. supports both advanced generic drug programs and complex biosimilar development from its modern analytical facility located in Laval, Quebec, Canada.
Operating under a fully implemented ISO 9001:2015 certified Quality Management System, USFDA registration (Establishment Identifier 3042696771), and Health Canada Drug Establishment Licence 3-002945-A, ResolveMass Laboratories maintains exceptional standards of data integrity, analytical quality, and regulatory compliance. By leveraging advanced LC-MS/MS, GC-MS, high-field NMR, and optimized protein digestion workflows, our team of PhD-level scientists delivers the comprehensive molecular characterization required to satisfy today’s increasingly rigorous global regulatory expectations.
For customized analytical testing, formulation development, biosimilar characterization, or regulatory support services, please contact our scientific team directly.
Contact ResolveMass Laboratories Inc.
Frequently Asked Questions (FAQs)
The manufacturing approach depends on the nature of the active ingredient. Generic drugs are produced through controlled chemical synthesis, enabling the creation of identical molecular structures in every batch. Biosimilars, however, are manufactured using genetically engineered living cells that naturally produce complex proteins. Since biological systems introduce inherent variability, advanced process controls and analytical characterization are essential to ensure product consistency.
The phrase “highly similar” means that a biosimilar closely matches its reference biologic in structural characteristics, biological activity, purity, and clinical performance. Regulatory authorities evaluate multiple critical quality attributes, including the primary amino acid sequence, higher-order structure, glycosylation profile, and functional activity. Any minor differences identified must not affect the product’s safety, efficacy, or quality.
An Abbreviated New Drug Application (ANDA) is used for generic drugs and primarily requires evidence of chemical equivalence and pharmacokinetic bioequivalence with the reference product. A 351(k) biosimilar application follows a more comprehensive pathway that includes extensive analytical characterization, functional testing, comparative pharmacokinetic studies, immunogenicity assessments, and a totality-of-the-evidence approach to establish biosimilarity.
Not necessarily. Current regulatory guidance from agencies such as Health Canada and the US FDA places greater emphasis on advanced analytical and functional comparability studies. When a biosimilar demonstrates a high degree of similarity through comprehensive structural characterization, comparative pharmacokinetic evaluations, and immunogenicity assessments, large Phase III comparative efficacy studies are often no longer required.
An interchangeable biosimilar is a biosimilar that satisfies additional regulatory requirements demonstrating that patients can safely switch between it and the reference biologic without increased risk or reduced effectiveness. In jurisdictions where interchangeability is recognized, pharmacists may substitute the interchangeable product without obtaining approval from the prescribing physician, provided local regulations permit automatic substitution.
Peptide mapping is a key analytical technique for confirming that a biosimilar has the same primary amino acid sequence as its reference biologic. Using high-resolution LC-MS/MS, scientists can detect sequence variants, identify post-translational modifications, and monitor degradation-related changes such as oxidation or deamidation. This detailed molecular information is essential for demonstrating structural comparability during regulatory submissions.
Biosimilar development requires substantially greater investment because biologics are produced using complex living-cell manufacturing systems and demand extensive analytical characterization throughout development. In addition to cell-line development and large-scale bioprocessing, manufacturers must perform comprehensive structural, functional, pharmacokinetic, and immunogenicity studies. Generic drugs, by comparison, rely mainly on chemical synthesis and bioequivalence testing, resulting in much lower development costs.
Reference:
- U.S. Food and Drug Administration. (2022). Considerations in demonstrating interchangeability with a reference product: Update (Guidance for Industry). https://www.fda.gov/media/154912/download
- Pagani, E. (2019). Why are biosimilars much more complex than generics? Einstein (São Paulo), 17(1), eED4836. https://doi.org/10.31744/einstein_journal/2019ED4836
- Triplitt, C., Hinnen, D., & Valentine, V. (2017). How similar are biosimilars? What do clinicians need to know about biosimilar and follow-on insulins? Clinical Diabetes, 35(4), 209–216. https://doi.org/10.2337/cd16-0072
- Nupur, N., Joshi, S., Gulliarme, D., & Rathore, A. S. (2022). Analytical similarity assessment of biosimilars: Global regulatory landscape, recent studies and major advancements in orthogonal platforms. Frontiers in Bioengineering and Biotechnology, 10, 832059. https://doi.org/10.3389/fbioe.2022.832059
- Health Canada. (2026, May 21). Summary of changes: Guidance on information and submission requirements for biosimilar biologic drugs. Government of Canada. https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/applications-submissions/guidance-documents/information-submission-requirements-biosimilar-biologic-drugs/summary-changes.html
- Mora, M., & González, C. (2015). Biosimilar: What it is not. British Journal of Clinical Pharmacology, 80(5), 949–956. https://doi.org/10.1111/bcp.12656
- European Medicines Agency. (2023, April 21). Q&A on the statement on the scientific rationale supporting interchangeability of biosimilar medicines in the EU (EMA/93740/2023 Rev. 1). https://www.ema.europa.eu/en/documents/other/qa-statement-scientific-rationale-supporting-interchangeability-biosimilar-medicines-eu_en.pdf
- Health Canada. (2026, May 19). Biosimilar biologic drugs in Canada: Fact sheet. Government of Canada. https://www.canada.ca/en/health-canada/services/drugs-health-products/biologics-radiopharmaceuticals-genetic-therapies/applications-submissions/guidance-documents/fact-sheet-biosimilars.html

