
Introduction:
Setting a therapeutic peptide API purity specification is one of the most consequential — and most frequently misjudged — decisions in a peptide drug development program. Sponsors often default to copying a purity number from a compendial monograph or a competitor’s filing, only to face FDA or EMA information requests asking them to justify why that number is appropriate for their specific peptide, synthesis route, and impurity profile. Because peptide APIs sit in a regulatory gray zone between small-molecule drug substances and biologics, the specification-setting logic in ICH Q3A(R2) and ICH Q6A has to be adapted rather than applied verbatim, and reviewers expect that adaptation to be documented, not assumed.
This article breaks down what purity range is realistically achievable and defensible for synthetic peptide APIs, how impurity thresholds should be structured, which impurity classes draw the closest scrutiny, and how the expectations differ between NDA and ANDA pathways. It also covers the analytical techniques — sequencing, peptide mapping, LC-MS, and related orthogonal methods — that regulators expect to see behind the purity number itself, with particular attention to GLP-1 peptide APIs, where characterization and comparability expectations have become especially rigorous.
Summary:
- An acceptable therapeutic peptide API purity specification is not a single fixed number — it is risk-based, driven by the peptide’s synthesis route, molecular size, and clinical dose, and must be justified against ICH Q6A/Q3A(R2) principles and batch history data.
- Most regulatory-accepted specifications for synthetic peptide APIs fall between 95.0% and 99.0% purity by RP-HPLC (area normalization), with individual unspecified impurities typically capped at 0.10%–0.50% depending on peptide length and dose.
- FDA and EMA both expect impurity identification thresholds, qualification thresholds, and reporting thresholds to be set using ICH Q3A(R2)-derived logic adapted for peptides, since ICH Q3A was written for small molecules, not peptide APIs.
- ANDA submissions for peptide generics carry the added burden of demonstrating impurity profile equivalence to the Reference Listed Drug (RLD), not just meeting a generic purity cutoff.
- Deamidation, oxidation, diastereomers, truncated sequences, and aggregation products are the impurity classes regulators scrutinize most closely in peptide APIs.
- Orthogonal sequencing, peptide mapping, and mass spectrometry techniques are what actually confirm purity claims, especially for complex analogs like GLP-1 peptides.
- A well-documented, forced-degradation-supported specification — not just a tight number — is what actually satisfies FDA/EMA reviewers and prevents Complete Response Letters (CRLs) tied to CMC deficiencies.
1: Why “Acceptable Purity” Is a Moving Target for Peptide APIs
There is no universal purity number that regulators accept for every therapeutic peptide API — the acceptable therapeutic peptide API purity specification depends on the peptide’s synthesis complexity, chain length, and the sensitivity of its impurities to patient safety. A 9-residue peptide like octreotide and a 39-residue peptide like liraglutide do not carry the same realistic purity ceiling, and regulators know this.
This is precisely why ICH Q6A allows specifications to be set based on manufacturing experience and batch data rather than an arbitrary compendial value. For peptide APIs specifically, both FDA’s Office of Pharmaceutical Quality and EMA’s Quality Working Party expect sponsors to justify their purity limit with development history, not simply propose a number that “looks good” on paper.
2: What Purity Range Do FDA and EMA Actually Accept?
Most approved synthetic peptide APIs carry a purity specification of 95.0%–99.0% by RP-HPLC, though the exact figure depends on the synthesis route (solid-phase vs. liquid-phase vs. hybrid) and the peptide’s structural complexity.
| Peptide Complexity | Typical Purity Specification | Common Analytical Method |
|---|---|---|
| Short linear peptides (≤10 residues) | 98.0%–99.5% | RP-HPLC, UV 214–220 nm |
| Mid-length peptides (11–25 residues) | 96.0%–98.5% | RP-HPLC with orthogonal CE or UPLC |
| Long/complex peptides (26–50 residues) | 95.0%–97.5% | RP-HPLC + LC-MS confirmation |
| Cyclic or disulfide-bridged peptides | 94.0%–97.0% (case-specific) | RP-HPLC + peptide mapping |
These ranges are directional, not mandates — a sponsor’s actual specification must trace back to clinical batch data, stability trends, and process capability, consistent with ICH Q6A general specification-setting principles. For cyclic and disulfide-constrained molecules in particular, sponsors typically need a dedicated cyclic peptide characterization workflow, since ring closure and disulfide scrambling impurities are not always resolved by standard linear-peptide methods.
3: What Impurity Thresholds Apply to Peptide APIs?
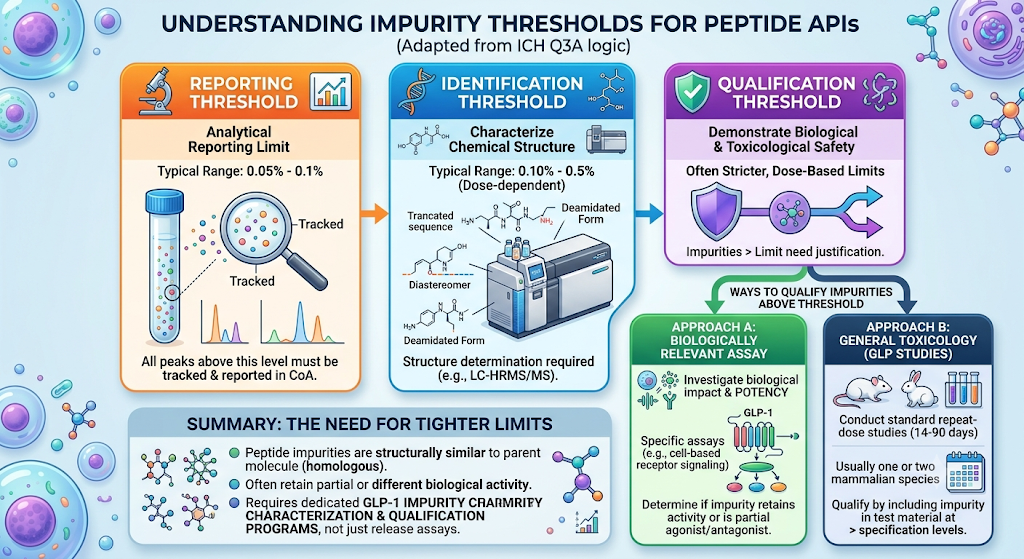
Peptide API impurities are governed by adapted ICH Q3A(R2) logic, since the original guideline was designed for small-molecule drug substances, not synthetic or recombinant peptides. Reviewers typically expect three threshold tiers to be defined and justified in the specification:
- Reporting Threshold — the level above which an impurity peak must be reported, generally 0.05%–0.1% for peptide APIs.
- Identification Threshold — the level above which an impurity’s structure must be characterized (often via LC-MS/MS), commonly set at 0.10%–0.5% depending on maximum daily dose.
- Qualification Threshold — the level requiring toxicological or biological qualification, frequently tied to dose-based limits rather than a flat percentage.
Because peptide-related impurities (deamidated forms, diastereomers, truncated sequences) are often structurally similar to the parent peptide and may retain partial biological activity, regulators frequently ask for tighter identification and qualification thresholds than would apply to a comparable small-molecule impurity at the same percentage level. This is one of the main reasons sponsors invest in dedicated GLP-1 peptide impurity characterization programs rather than relying on a single release assay to carry the whole impurity story.
3: What Impurity Thresholds Apply to Peptide APIs?
Peptide API impurities are governed by adapted ICH Q3A(R2) logic, since the original guideline was designed for small-molecule drug substances, not synthetic or recombinant peptides. Reviewers typically expect three threshold tiers to be defined and justified in the specification:
- Reporting Threshold — the level above which an impurity peak must be reported, generally 0.05%–0.1% for peptide APIs.
- Identification Threshold — the level above which an impurity’s structure must be characterized (often via LC-MS/MS), commonly set at 0.10%–0.5% depending on maximum daily dose.
- Qualification Threshold — the level requiring toxicological or biological qualification, frequently tied to dose-based limits rather than a flat percentage.
Because peptide-related impurities (deamidated forms, diastereomers, truncated sequences) are often structurally similar to the parent peptide and may retain partial biological activity, regulators frequently ask for tighter identification and qualification thresholds than would apply to a comparable small-molecule impurity at the same percentage level. This is one of the main reasons sponsors invest in dedicated GLP-1 peptide impurity characterization programs rather than relying on a single release assay to carry the whole impurity story.
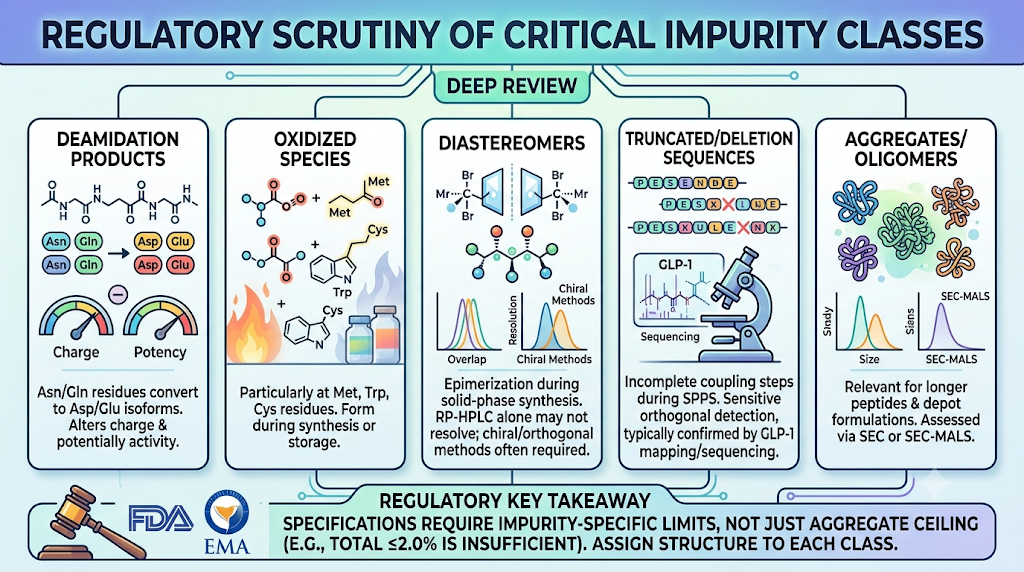
4: Which Impurity Classes Get the Most Regulatory Scrutiny?
Deamidation products, oxidized variants, diastereomers, truncated/deletion sequences, and aggregates draw the closest FDA and EMA review because each can affect potency, immunogenicity, or safety differently than a simple process-related impurity.
- Deamidation products — Asn/Gln residues converting to Asp/Glu isoforms, altering charge and potentially activity.
- Oxidized species — particularly at Met, Trp, and Cys residues, often forming during synthesis or storage.
- Diastereomers — epimerization during solid-phase synthesis, which RP-HPLC alone may not resolve; chiral or orthogonal methods are often required.
- Truncated and deletion sequences — incomplete coupling steps during SPPS, requiring sensitive orthogonal detection, typically confirmed through GLP-1 impurity peptide mapping.
- Aggregates/oligomers — more relevant for longer peptides and depot formulations, assessed via SEC or SEC-MALS.
A specification that only reports “total impurities ≤2.0%” without breaking out these classes individually is unlikely to satisfy a CMC reviewer — regulators expect impurity-specific limits, not just an aggregate ceiling. For GLP-1 analogs specifically, sponsors increasingly rely on GLP-1 peptide impurity sequencing analysis to assign structure to each impurity class before setting individual limits.
5: How Sequencing and Mapping Confirm Purity and Identity
A purity number by itself does not prove a peptide API is what the sponsor claims it is — sequence confirmation through peptide sequencing and peptide mapping is what ties the purity result to a verified molecular identity.
For GLP-1 analogs, this identity confirmation typically combines several complementary techniques rather than one method carrying the full burden of proof:
| Technique | What It Confirms | Typical Use Case |
|---|---|---|
| LC-MS characterization | Mass confirmation, impurity mass assignment | Routine identity and impurity structure elucidation |
| HRMS peptide mapping | High-resolution fragment-level sequence confirmation | Detecting low-level sequence variants |
| Enzymatic digestion mapping | Site-specific sequence coverage | Confirming full-length sequence integrity |
| De novo sequencing | Sequence confirmation without reference database bias | Generic/biosimilar sequence verification |
| Native mass spectrometry | Higher-order structure and non-covalent interactions | Complex or self-associating peptide APIs |
| Peptide mapping vs. intact mass analysis | Comparative confidence in identity confirmation | Choosing the right primary confirmatory method |
For semaglutide specifically, semaglutide peptide mapping has become a standard reference workflow that sponsors adapt when characterizing other GLP-1 analogs, given the overlapping impurity and degradation pathways across the class. Reviewers are also increasingly interested in GLP-1 sequence variant analysis, which distinguishes true sequence-level impurities from post-translational or synthesis-related modifications that only look like sequence variants on a chromatogram.
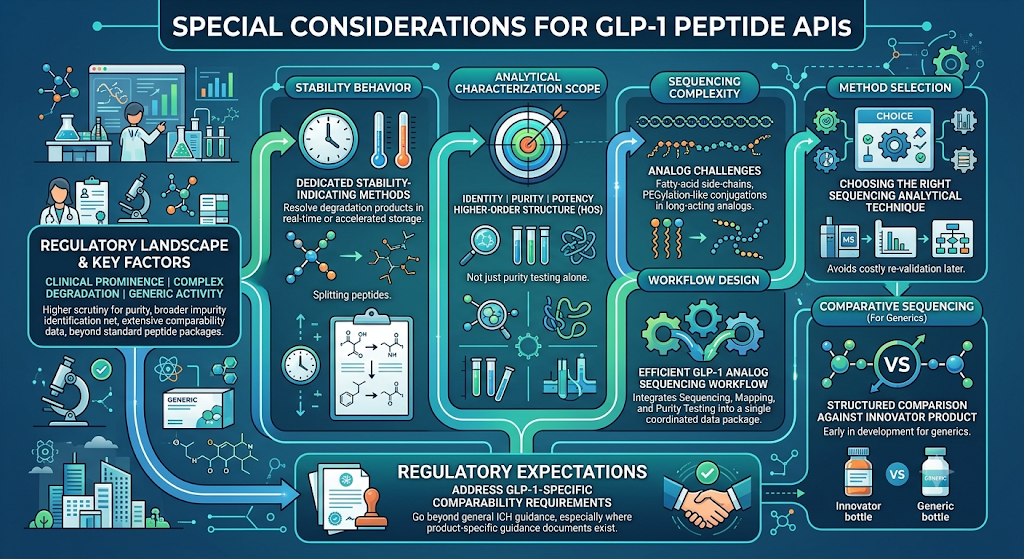
6: Special Considerations for GLP-1 Peptide APIs
GLP-1 peptide APIs face additional purity and characterization scrutiny because of their clinical prominence, complex degradation pathways, and the volume of generic development activity currently underway. The regulatory requirements for GLP-1 peptide characterization go beyond a standard peptide API package, often requiring a broader impurity identification net and more extensive comparability data than older, simpler peptide APIs.
Key GLP-1-specific factors sponsors need to plan for include:
- Stability behavior — GLP-1 analogs often require dedicated stability-indicating analytical methods capable of resolving degradation products that emerge only under real-time or accelerated storage conditions.
- Analytical characterization scope — a complete analytical characterization of GLP-1 peptide drugs package typically spans identity, purity, potency, and higher-order structure, not purity testing in isolation.
- Sequencing complexity — GLP-1 peptide sequencing challenges commonly arise from fatty-acid side-chain modifications and PEGylation-like conjugation strategies used in long-acting analogs.
- Method selection — choosing the right GLP-1 peptide sequencing analytical technique up front avoids costly re-validation later in development.
- Workflow design — an efficient GLP-1 analog peptide sequencing workflow integrates sequencing, mapping, and purity testing into a single coordinated data package rather than three disconnected studies.
- Comparative sequencing — sponsors developing generics benefit from a structured peptide sequencing of GLP-1 drugs comparison against the innovator product early in development.
Regulators reviewing GLP-1 filings also expect sponsors to address the GLP-1 peptide characterization regulatory requirements specific to comparability, not just general ICH peptide guidance, particularly where a product-specific guidance document exists.
7: Multi-Attribute Monitoring and Advanced Purity Confirmation
Beyond conventional RP-HPLC purity testing, many sponsors now supplement their specification package with multi-attribute monitoring (MAM), a mass spectrometry-based approach that monitors multiple quality attributes — including several impurity classes — within a single analytical run rather than relying on separate methods for each attribute.
MAM is particularly useful for peptide APIs with several co-occurring impurity types, since it can simultaneously track deamidation, oxidation, and sequence variants without requiring a separate validated method for each one. While MAM is not yet a mandatory expectation for every ANDA or NDA peptide filing, its use strengthens the overall impurity characterization narrative and is increasingly referenced favorably in regulatory interactions.
8: How Is Purity Different for ANDA vs. NDA Peptide Submissions?
An NDA sponsor sets the purity specification based on its own clinical and stability data, while an ANDA sponsor must additionally demonstrate that its peptide API’s impurity profile is qualitatively and quantitatively comparable to the Reference Listed Drug’s approved profile.
| Requirement | NDA Pathway | ANDA Pathway (Peptide Generic) |
|---|---|---|
| Basis for purity limit | Clinical batch/stability data | Comparative data vs. RLD impurity profile |
| New impurities above RLD levels | Allowed with toxicological qualification | Generally requires additional qualification/justification |
| Reference standard | Sponsor-developed | Often cross-referenced or independently qualified against RLD |
| FDA guidance basis | ICH Q6A, Q3A(R2) | Same, plus product-specific guidance (PSG) where available |
For several synthetic peptide generics (e.g., glucagon, teriparatide, glatiramer acetate), FDA has issued product-specific guidances that define expected physicochemical and impurity comparability criteria — these should always be checked before finalizing a purity specification for an ANDA filing.
9: What Documentation Actually Supports the Specification?
A purity specification is only as strong as the data package behind it — regulators expect forced degradation studies, multiple qualification batches, and orthogonal method verification, not just a single validated HPLC method.
- Forced degradation studies (acid, base, oxidative, thermal, photolytic) to demonstrate the method’s stability-indicating capability.
- Batch-to-batch trending data across development and validation lots to justify the proposed range statistically.
- Orthogonal analytical confirmation (e.g., CE, UPLC, LC-MS/MS) to catch co-eluting impurities RP-HPLC alone might miss.
- Reference standard characterization with full impurity profiling, not just an assay value.
10: Why Sponsors Outsource Peptide Purity and Impurity Characterization
Building an in-house program capable of covering purity, sequencing, mapping, and impurity characterization for a peptide API is resource-intensive, which is why most sponsors work with a specialized CRO for GLP-1 peptide characterization rather than assembling every capability internally.
Before selecting a partner, sponsors should be clear on two things: what deliverables they actually need, and what information the CRO needs from them to scope the work accurately.
- Review a peptide characterization CRO deliverables checklist to confirm the scope covers identity, purity, impurity structure elucidation, and stability — not just a single assay report.
- Understand the specifications to provide when outsourcing peptide characterization to a CRO, including synthesis route, known impurity history, and intended regulatory pathway, so the study design matches the filing strategy from the start.
- For sequencing-specific work, sponsors can engage GLP-1 peptide sequencing CRO services as a standalone workstream or bundled with broader characterization.
- Many sponsors specifically choose to outsource GLP-1 peptide sequencing services when internal MS capacity or peptide-specific method expertise is limited.
Conclusion:
There is no single “safe” number when it comes to a therapeutic peptide API purity specification — an acceptable limit is one supported by synthesis-appropriate analytical methods, forced degradation data, sequence and mapping confirmation, and (for ANDAs) direct comparability to the Reference Listed Drug’s impurity profile. Sponsors who treat the purity specification as a checkbox rather than a documented, risk-based conclusion are the ones most likely to face CMC-related review questions or a Complete Response Letter.
ResolveMass Laboratories supports pharmaceutical sponsors with peptide API purity method development, forced degradation studies, sequencing, peptide mapping, impurity characterization, and ANDA/NDA-ready CMC documentation packages built on ICH-aligned, regulator-tested approaches.
Frequently Asked Questions:
There is no single FDA or ICH requirement that applies to every peptide API. However, commercial therapeutic peptide APIs are commonly expected to demonstrate purity levels of 95–98% or higher, depending on the molecule and intended use. Regulatory authorities assess whether the proposed specification is supported by scientific evidence rather than focusing on a fixed percentage. Factors such as impurity profile, manufacturing capability, clinical batch comparability, and stability data influence the acceptable limit. Each specification should be justified using validated analytical methods and comprehensive characterization.
Impurity profiling is essential because even low levels of impurities can affect the safety, efficacy, stability, or immunogenicity of therapeutic peptides. It helps identify both product-related and process-related impurities generated during synthesis or storage. Comprehensive impurity profiling demonstrates that the manufacturing process is well controlled and consistently produces high-quality material. Regulatory agencies expect manufacturers to identify, quantify, and evaluate impurities using validated analytical methods. Proper impurity control reduces regulatory concerns and supports successful ANDA and NDA submissions.
Impurity limits are established by evaluating manufacturing process capability, analytical characterization, toxicological data, stability studies, and clinical batch information. Manufacturers assess whether impurities remain within safe and acceptable levels throughout the product’s shelf life. Each proposed limit should be supported by scientific evidence and validated analytical methods. Regulatory authorities also consider the consistency of multiple manufacturing batches and the potential impact of impurities on product quality. A risk-based approach ensures that impurity specifications are appropriate for both patient safety and product performance.
Product-related impurities originate from changes in the peptide molecule itself, such as truncated sequences, oxidized variants, deamidated peptides, or aggregation products. Process-related impurities are introduced during manufacturing and may include residual solvents, coupling reagents, protecting groups, catalysts, resin-derived contaminants, or counter ions. Both types of impurities must be identified, characterized, and controlled because they can influence product quality and regulatory compliance. Comprehensive impurity profiling helps manufacturers establish robust specifications and maintain consistent manufacturing processes.
The CMC section should include the proposed purity specification, analytical methods, validation reports, impurity profiles, batch analysis data, stability study results, and scientific justification for acceptance criteria. Manufacturers should also describe the manufacturing process, demonstrate batch-to-batch consistency, and provide evidence that analytical methods are suitable for their intended purpose. Comprehensive documentation helps regulators assess whether the peptide API consistently meets quality standards. A well-prepared CMC package reduces review questions and facilitates smoother regulatory approval.
Reference
- Xu J. Peptides chemistry, manufacturing, and controls. Peptide science: chemical ligation, lead generation, and therapeutic advances. 2025 Jul 4:471-508.https://onlinelibrary.wiley.com/doi/abs/10.1002/9781119824701.ch14
- Rastogi S, Shukla S, Kalaivani M, Singh GN. Peptide-based therapeutics: quality specifications, regulatory considerations, and prospects. Drug Discovery Today. 2019 Jan 1;24(1):148-62.https://www.sciencedirect.com/science/article/pii/S1359644618302514
- Jois SD. Regulatory Issues for Peptide Drugs. InPeptide Therapeutics: Fundamentals of Design, Development, and Delivery 2022 Sep 27 (pp. 287-305). Cham: Springer International Publishing.https://link.springer.com/chapter/10.1007/978-3-031-04544-8_9
- Srivastava V, editor. Peptide therapeutics: strategy and tactics for chemistry, manufacturing, and controls. Royal Society of Chemistry; 2019 Aug 16.https://books.google.com/books?hl=en&lr=&id=Cw6xDwAAQBAJ&oi=fnd&pg=PT20&dq=What+Purity+Specification+Is+Acceptable+for+a+Therapeutic+Peptide+API+in+an+ANDA+or+NDA+Submission%3F&ots=BTDbfqYeN0&sig=zWmnba5ah1eILWttlZmF1pXLJhs
- Van Dorpe S, Verbeken M, Wynendaele E, De Spiegeleer B. Purity profiling of peptide drugs. JOURNAL OF BIOANALYSIS AND BIOMEDICINE. 2011(S6).https://www.researchgate.net/profile/Bart-De-Spiegeleer/publication/273797539_Purity_profiling_of_Peptide_Drugs/links/571a472c08ae408367bc87d6/Purity-profiling-of-Peptide-Drugs.pdf

