
Introduction to Extractables and Leachables Thresholds in Impurity Control
Extractables and leachables thresholds are scientifically established safety limits used to determine whether migrating chemical compounds must be reported, structurally identified, and toxicologically qualified. By defining clear exposure-based cutoffs according to patient dosage and route of administration, these thresholds enable manufacturers to concentrate analytical efforts on compounds that may present a meaningful clinical risk.
Throughout the manufacturing, storage, and administration of pharmaceutical products and biologics, drug formulations inevitably come into direct contact with process equipment, container-closure systems, and drug-delivery devices. These contact materials include elastomeric seals, polymeric containers, administration tubing, and single-use processing systems that contain low-molecular-weight additives, plasticizers, antioxidants, stabilizers, and vulcanization agents. Under manufacturing stress conditions or during prolonged storage, these substances may migrate from the material matrix into the drug product. This migration is categorized into two distinct analytical groups: extractables, which are released under aggressive laboratory conditions to establish a worst-case risk profile, and leachables, which migrate into the therapeutic product under normal storage and usage conditions.
Assessing these substances through established Extractables and Leachables Thresholds is critical because uncontrolled chemical migration may affect drug stability, compromise active pharmaceutical ingredients, or expose patients directly to potentially harmful compounds. To minimize these risks, regulatory authorities apply stringent requirements outlined in pharmacopoeial standards such as USP <1663> and USP <1664>, ISO 10993 guidance documents, and the draft ICH Q3E guideline.
Need to optimize your testing budget? Understand the true cost of extractables and leachables testing.
Share via:
Article Summary:
- Extractables and leachables (E&L) are chemical substances that can migrate from packaging materials, manufacturing equipment, or delivery devices into pharmaceutical products. Their assessment is essential to ensure product quality, stability, and patient safety.
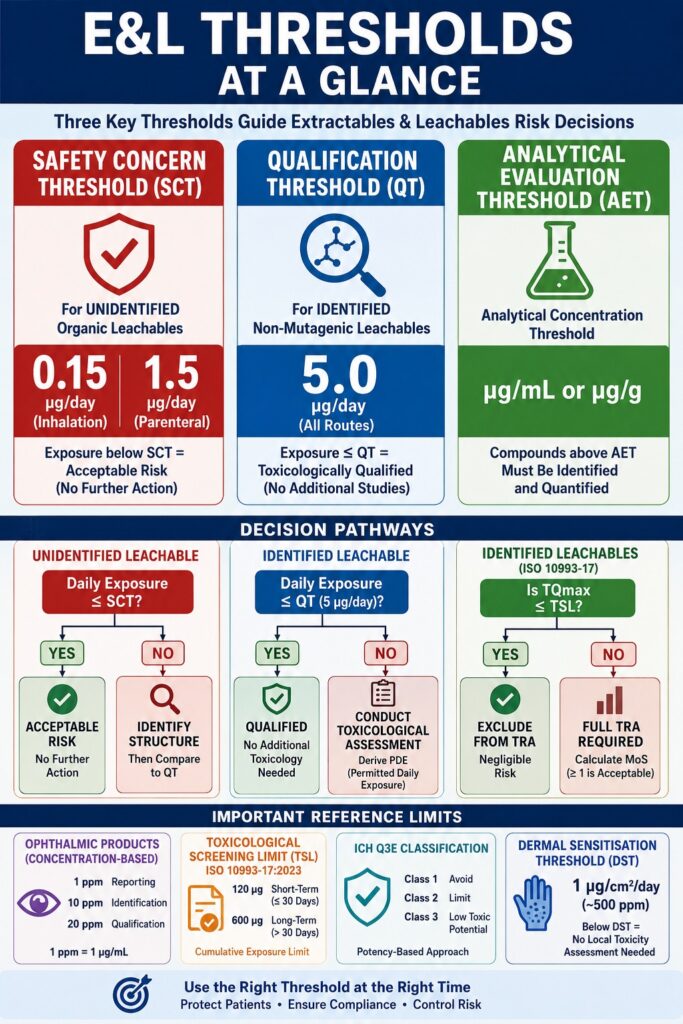
- Three core thresholds guide E&L risk assessment: the Safety Concern Threshold (SCT) for unidentified compounds, the Qualification Threshold (QT) for identified non-mutagenic compounds, and the Analytical Evaluation Threshold (AET) used to establish laboratory testing limits.
- SCT defines acceptable exposure levels for unidentified leachables. Exposure below the SCT is generally considered to present negligible health risk, while compounds exceeding the threshold require identification and further evaluation.
- QT serves as a toxicological decision point. Identified non-mutagenic leachables with exposures below the QT typically require no additional safety studies, whereas higher exposures may trigger compound-specific toxicological assessments and derivation of a Permitted Daily Exposure (PDE).
- AET converts toxicological limits into measurable analytical targets. It helps laboratories determine which detected compounds must be identified and quantified, ensuring analytical sensitivity remains aligned with patient exposure risk.
- Additional regulatory concepts enhance risk management, including Uncertainty Factors (UF) to account for analytical variability, concentration-based thresholds for ophthalmic products, and the Toxicological Screening Limit (TSL) for medical devices to streamline evaluation of low-risk compounds.
- Emerging global guidance, including draft ICH Q3E, promotes a risk-based classification approach for leachables, helping manufacturers prioritize control strategies, improve regulatory compliance, and focus resources on compounds with the greatest potential impact on patient safety.
Extractables and Leachables Thresholds: Defining the Core Concepts of SCT, QT, and AET
The principal parameters used within extractables and leachables threshold assessments are the Safety Concern Threshold (SCT), the Qualification Threshold (QT), and the Analytical Evaluation Threshold (AET). While the SCT and QT are toxicological exposure limits expressed in micrograms per day, the AET is an analytical concentration threshold that translates those dose-based limits into measurable concentrations suitable for laboratory testing.
Historically, chemical evaluations of packaging materials generated extensive datasets containing numerous detected compounds. However, there was often no scientifically justified framework for determining which trace-level compounds required additional evaluation. The adoption of threshold-based methodologies resolved this challenge by creating clear decision criteria. If a migrating compound is present below established safety thresholds, the associated health risk is considered negligible, and additional identification or qualification activities are generally unnecessary.
This systematic approach aligns analytical sensitivity with toxicological relevance. Instead of relying solely on arbitrary instrumental limits of detection (LOD), which may differ among laboratories, risk assessments are based on actual patient exposure. Modern extractables and leachables programs rely on these thresholds to direct screening studies, structural characterization, and toxicological qualification activities.
| Threshold Parameter | Expressed Unit | Toxicological or Analytical Objective | Primary Regulatory Context |
|---|---|---|---|
| Safety Concern Threshold (SCT) | μg/day (Total Daily Intake) | Establishes the exposure level below which an unidentified leachable presents negligible mutagenic or non-mutagenic risk. | PQRI, USP <1664>, draft ICH Q3E |
| Qualification Threshold (QT) | μg/day (Total Daily Intake) | Establishes the exposure level below which an identified non-mutagenic leachable does not require formal toxicological qualification. | PQRI, USP <1664>, draft ICH Q3E |
| Analytical Evaluation Threshold (AET) | μg/mL or μg/g | Represents the concentration threshold above which compounds must be identified and quantified. | ISO 10993-18, USP <1663>, draft ICH Q3E |
Addressing specialized delivery systems? Explore E&L testing for pre-filled syringes
and extractables and leachables testing for autoinjectors.
The Safety Concern Threshold: Scientific Derivation and Application
The Safety Concern Threshold (SCT) represents the maximum daily patient exposure level below which an individual organic leachable is considered to pose a negligible risk of carcinogenic and non-carcinogenic toxicity. Based on quantitative risk assessment methodologies, the SCT is established at 0.15 μg/day for inhalation drug products and 1.5 μg/day for parenteral drug products.
The scientific basis for the SCT originated from the work of the Product Quality Research Institute (PQRI) Leachables and Extractables Working Group. This group integrated extensive toxicological and chemical databases to address risks associated with container-closure system migration. To derive the default SCT value of 0.15 μg/day for Orally Inhaled and Nasal Drug Products (OINDP), the working group evaluated Risk Specific Doses obtained from genotoxic carcinogen studies within the Carcinogenic Potency Database (CPDB). Using a conservative lifetime excess cancer risk level of 1 × 10⁻⁶ (one additional cancer case per million individuals) and assuming a standard human body weight of 50 kg, the resulting limit of 0.15 μg/day was established as a level at which both carcinogenic and non-carcinogenic risks are considered negligible.
Are you developing respiratory therapeutics? Learn more about E&L testing for inhalation and nasal drug products.
For Parenteral Drug Products (PDP), administered through intravenous, subcutaneous, or intramuscular routes, the PQRI Parenteral and Ophthalmic Drug Product (PODP) Working Group extended these risk assessment principles. Since parenteral formulations are often aqueous and generally less likely to extract highly potent hydrophobic carcinogens, a lifetime excess cancer risk level of 1 × 10⁻⁵ is considered acceptable. This risk level corresponds to a standard SCT value of 1.5 μg/day for parenteral products.
Decision Process for Unidentified Organic Leachables
Unidentified Organic Leachable
│
├─► Daily Exposure ≤ SCT? ──► Yes ──► No Further Action Required (Acceptable Risk)
│
└─► Daily Exposure > SCT? ──► No ───► Identify Structure and Compare to QT
Standard SCT values apply to unidentified organic compounds under the assumption of a conservative worst-case toxicological profile. However, highly potent carcinogenic subclasses known collectively as the “cohort of concern,” including N-nitrosamines, polycyclic aromatic hydrocarbons (PAHs), and aflatoxin-like compounds, are excluded from default SCT assumptions and require compound-specific acceptable intake limits.
The Qualification Threshold: When Is Toxicological Evaluation Required?
The Qualification Threshold (QT) is the exposure level below which an identified non-carcinogenic leachable does not require formal toxicological qualification. Established at 5 μg/day for both inhalation and parenteral administration routes, the QT serves as a regulatory trigger for compound-specific hazard assessment whenever this limit is exceeded.
The QT functions as a practical decision-making filter within safety qualification programs. When analytical screening identifies a leachable at levels above the SCT, investigators must determine its molecular structure to perform a toxicological risk assessment. If the compound is identified and does not contain structural alerts associated with genotoxic carcinogenicity, reproductive toxicity, sensitization, or local irritation, its estimated patient exposure is compared with the 5 μg/day QT. If exposure remains at or below this threshold, the compound is considered toxicologically qualified for its intended clinical application, and no additional safety studies are typically required.
When an identified leachable exceeds the 5 μg/day threshold, a formal safety qualification process becomes necessary. This process requires a compound-specific risk assessment to establish a Permitted Daily Exposure (PDE). Toxicologists review available in vivo and in vitro data, determine an appropriate Point of Departure (such as the No Observed Adverse Effect Level [NOAEL], Lowest Observed Adverse Effect Level [LOAEL], or Benchmark Dose Lower Confidence Limit [BMDL]), and apply relevant uncertainty factors to derive a clinically acceptable exposure limit. If adequate toxicological information is unavailable, additional studies, including genotoxicity testing or general toxicity evaluations, may be required to demonstrate patient safety.
| Route of Administration | Daily Exposure Limit (Mutagenic) | Daily Exposure Limit (Non-Mutagenic) | Core Reference Standard |
| Orally Inhaled (OINDP) | 0.15 μg/day (SCT) | 5.0 μg/day (QT) | PQRI Inhalation Recommendations, USP <1664.1> |
| Parenteral (PDP) | 1.5 μg/day (SCT) | 5.0 μg/day (QT) | PQRI Parenteral Recommendations, draft ICH Q3E |
| Topical Ophthalmic | 1 ppm (Reporting) | 20 ppm (Qualification) | FDA Draft Quality Considerations for Topical Ophthalmic Products |
Need help with compliance? Get expert guidance on the toxicological qualification of leachables.
Establishing the Analytical Evaluation Threshold: Formulas and Derivations
The Analytical Evaluation Threshold (AET) is mathematically derived to convert dose-based toxicological limits into concentration-based analytical targets suitable for laboratory testing. By relating patient exposure limits to extraction volumes and material characteristics, the AET establishes the minimum sensitivity required for extractables and leachables screening methods.
To apply the AET concept, laboratories convert a dose-based patient exposure limit into an instrumental concentration threshold. For pharmaceutical packaging systems and single-use manufacturing components, the unadjusted initial AET (AETinitial in μg/mL) is calculated using the following equation:
AETinitial = (SCT ÷ MDD) × (Vextract ÷ (Vcontact × f))
Where:
SCT = Safety Concern Threshold (μg/day), typically 1.5 μg/day for parenteral products or 0.15 μg/day for inhalation products.
MDD = Maximum Daily Dose, representing the maximum volume of drug product administered during a 24-hour period (mL/day).
Vextract = Total extraction solvent volume used during laboratory extraction studies (mL).
Vcontact = Physical volume or contact surface area of the packaging or device component (mL or cm²).
f = Accumulation factor accounting for cumulative exposure from multiple equivalent components.
For drug-device combinations and container-closure systems containing a fixed number of labeled doses, an alternative estimation equation (AETestimated in μg/g or μg/canister) is used:
AETestimated = (SCT ÷ Doses/day) × (DosesCCS ÷ WeightCCS)
Where:
Doses/day = Number of doses administered during a 24-hour period.
DosesCCS = Total number of doses contained within a single container-closure system.
WeightCCS = Total mass, volume, or surface area of the contact material within the container-closure system.
Metered Dose Inhaler (MDI) Component Calculation
Consider a metered-dose inhaler containing 200 labeled actuations per canister, a maximum recommended daily usage of 12 actuations, and a valve elastomer weighing 200 mg (0.2 g). Using the inhalation SCT value of 0.15 μg/day:
AETestimated = (0.15 μg/day ÷ 12 actuations/day) × 200 actuations/canister
AETestimated = 2.5 μg/canister
To express this value relative to the elastomer mass:
AETcomponent = 2.5 μg/canister ÷ 0.2 g/canister
AETcomponent = 12.5 μg/g
Dry Powder Inhaler (DPI) Blister Calculation
Consider a dry powder inhaler blister containing 13 mg of formulation and 50 mg of contact foil, with a recommended dosing schedule of two actuations per day.
AETestimated = (0.15 μg/day ÷ 2 doses/day) × 1 dose/blister
AETestimated = 0.075 μg/blister
AETmaterial = 0.075 μg/blister ÷ 0.050 g/blister
AETmaterial = 1.5 μg/g
Curious about cost factors? View our guide on E&L testing cost.
The Crucial Role of the Analytical Uncertainty Factor in Chromatographic Screening
The analytical Uncertainty Factor (UF) is applied to calculated AET values to compensate for detector response variability during non-targeted screening studies. Its purpose is to reduce the likelihood of false-negative findings by ensuring that compounds with weak detector responses are not overlooked.
During non-targeted screening using gas chromatography-mass spectrometry (GC-MS) or liquid chromatography-mass spectrometry (LC-MS), unknown analytes are commonly quantified semi-quantitatively against an internal reference standard. This approach assumes that the unknown compound and the reference standard generate equivalent detector responses. In practice, however, detector relative response factors (RRFs) can vary substantially depending on molecular structure, ionization efficiency, and analytical detection mode.
If an analyte exhibits a low RRF relative to the internal standard, its concentration may be underestimated. Consequently, a potentially hazardous leachable could appear below the unadjusted AET, creating a false-negative (Type II) error. To address this risk, the initial AET is divided by an Uncertainty Factor to establish a more conservative adjusted AET:
AETadjusted = AETinitial ÷ UF
According to ISO 10993-18 and draft ICH Q3E recommendations, a default UF value of 2 is generally accepted for GC-MS methods because electron ionization typically provides relatively consistent detector responses. In contrast, LC-MS methods often exhibit greater variability due to differences in electrospray ionization efficiency. Therefore, LC-MS uncertainty factors should be derived empirically using representative relative response factor databases.
Relative Response Factor (RRF) Variability
Low RRF Variance (GC-MS / GC-FID)
↓
Default UF = 2
High RRF Variance (LC-MS)
↓
Empirical UF Derived from Database
UF = 1 ÷ (1 − RSD)
The statistical approaches recommended by ISO 10993-18 and PQRI utilize either standard deviation (Std) or relative standard deviation (RSD):
UF = 1 ÷ (1 − RSD)
UF = Mean ÷ [1 − (t × Std)]
Where:
Mean = Average relative response factor.
Std = Standard deviation of response factors.
t = Student’s t-value corresponding to the selected confidence level.
When response factor variability becomes excessive (RSD ≥ 1), the resulting UF approaches infinity, causing the adjusted AET to fall below the practical Limit of Detection (LOD). In such circumstances, the analytical method is considered unsuitable and must be optimized to improve response consistency.
Want to compare methodologies? Review the nuances of GC-MS vs LC-MS in extractables and leachables testing.
Concentration-Based Regulatory Limits: The Ophthalmic Exception
Concentration-based thresholds are used for topical ophthalmic drug products because the primary concern is local ocular toxicity rather than systemic exposure. Current FDA draft guidance establishes fixed thresholds consisting of a Reporting Threshold of 1 ppm, an Identification Threshold of 10 ppm, and a Qualification Threshold of 20 ppm.
For most drug administration routes, safety assessments are based on systemic exposure measured as total daily intake (μg/day). However, this approach does not adequately address local effects in ophthalmic products. Since ophthalmic drops deliver highly concentrated doses directly to a small area of ocular tissue, local concentration becomes the primary determinant of toxicity.
To address this unique exposure scenario, the FDA’s December 2023 Guidance for Industry on Quality Considerations for Topical Ophthalmic Drug Products established concentration-based limits expressed in parts per million (ppm), where 1 ppm equals 1 μg/mL. These thresholds replace traditional mass-based SCT and QT calculations and simplify analytical evaluation requirements for ophthalmic packaging systems.
| Threshold Type | Concentration Limit (ppm) | Required Laboratory Action | Primary Scientific Driver |
| Reporting Threshold | 1 ppm | Detect and document all migrant peak areas. | Establishes a baseline chemical profile. |
| Identification Threshold | 10 ppm | Determine molecular structures using mass spectrometric techniques. | Identifies compounds with potential local irritation concerns. |
| Qualification Threshold | 20 ppm | Conduct formal toxicological evaluations or safety assessments. | Limits exposure to concentrations considered safe for ocular tissues. |
The Toxicological Screening Limit: ISO 10993-17:2023 for Medical Devices
The Toxicological Screening Limit (TSL) is a cumulative exposure threshold introduced in ISO 10993-17:2023 that permits manufacturers to exclude low-level identified constituents from extensive toxicological assessment. The TSL is set at 120 μg for short-term exposure and 600 μg for long-term exposure, thereby streamlining biological evaluation activities for low-potency constituents.
Chemical characterization studies conducted under ISO 10993-18 frequently identify numerous migrating compounds. Performing comprehensive toxicological assessments on every detected constituent can be inefficient and may unnecessarily delay product development. To address this challenge, the 2023 revision of ISO 10993-17 formally incorporated the TSL as an exposure-based screening mechanism.
The TSL represents the cumulative exposure level below which risks associated with systemic toxicity, genotoxicity, carcinogenicity, and reproductive toxicity are considered negligible. The standard defines separate thresholds according to exposure duration:
Short-Term Exposure (≤ 30 days): 120 μg cumulative exposure limit.
Long-Term Exposure (> 30 days): 600 μg cumulative exposure limit.
To apply the TSL, manufacturers determine the maximum total quantity (TQmax) of the identified constituent. TQmax represents the total amount of chemical extractable from the device and is adjusted using a clinical scaling factor (SF) that reflects the maximum number of devices to which a patient may be exposed simultaneously.
TQmax = TQ × SF
If TQmax remains below the applicable TSL, the constituent is considered to present negligible toxicological risk and may be excluded from further evaluation.
TSL Decision Tree
Is the Chemical Structure Identified?
│
├──► No ───► Standard AET Evaluation Required
│
└──► Yes ──► Is It a Cohort of Concern?
│
├──► Yes ──► Apply Compound-Specific Limits
│
└──► No ───► Is TQmax ≤ TSL?
│
├──► Yes ──► Exclude from TRA
│
└──► No ──► Conduct Full TRA / MoS Assessment
The TSL cannot be applied to unidentified compounds because their structures must first be known to exclude membership within the cohort of concern. If TQmax exceeds the TSL, a formal Toxicological Risk Assessment (TRA) is required. This process involves calculating the Estimated Exposure Dose (EEDmax) and determining the Margin of Safety (MoS) relative to a Tolerable Intake (TI).
MoS = Tolerable Intake (TI) ÷ Worst-Case Estimated Exposure Dose (EEDmax)
An MoS value greater than or equal to 1 indicates that exposure remains below levels associated with adverse toxicological effects and is therefore considered acceptable.
Risk-Based Classification Under the Draft ICH Q3E Guideline
The draft ICH Q3E guideline introduces a potency-based classification system that categorizes organic leachables into three classes. This framework determines the extent of safety assessment and control measures required. Class 1 includes highly hazardous compounds that should be avoided whenever possible, Class 2 includes compounds managed using standard threshold concepts, and Class 3 includes low-toxicity compounds that do not require additional qualification when exposure remains below 1 mg/day.
Released in August 2025, the draft ICH Q3E guideline provides a globally harmonized and risk-based framework designed to reduce regional inconsistencies in extractables and leachables assessments. The guideline integrates quality risk management principles aligned with ICH Q9 and combines them with toxicological evaluation strategies to strengthen the control of both organic and inorganic impurities.
Class 1: Leachables to Be Avoided
This category includes mutagenic compounds belonging to the ICH M7 cohort of concern, substances with acceptable intake limits below 1.5 μg/day, and highly potent non-mutagenic toxicants such as bisphenol A and benzo[a]pyrene. Exposure must remain below compound-specific acceptable intake limits, and manufacturers should prioritize material replacement or process modifications to eliminate these compounds whenever possible.
Class 2: Leachables to Be Limited
This category represents the default classification for organic leachables where established Threshold of Toxicological Concern (TTC) and Qualification Threshold (QT) values are considered adequately protective. Exposure limits are route-specific and duration-specific and are generally derived from historical Permitted Daily Exposure (PDE) datasets.
Class 3: Leachables with Low Toxic Potential
This category includes compounds with well-documented low toxicity profiles, including mineral oil saturated hydrocarbons (MOSH) and synthetic polymer oligomers. Formal qualification is generally unnecessary when daily patient exposure remains below 1 mg/day.
To facilitate route-to-route extrapolation during Class 2 evaluations, the guideline introduces bioavailability correction factors (F6) based on oral bioavailability.
| Oral Bioavailability | Bioavailability Correction Factor (F6) | Application in PDE Derivation |
| < 1% | 100 | Used when extrapolating oral toxicity data to highly sensitive systemic exposure routes. |
| ≥ 1% to < 50% | 10 | Adjusts systemic exposure estimates for moderately absorbed compounds. |
| ≥ 50% to < 90% | 2 | Applied to compounds demonstrating substantial absorption. |
| ≥ 90% | 1 | Used when oral and systemic bioavailability are essentially equivalent. |
Additionally, for dermal and transdermal drug products, the guideline introduces a Dermal Sensitisation Threshold (DST) of 1 μg/cm²/day (approximately 500 ppm). Exposures below this threshold generally do not require formal local toxicity assessment, thereby simplifying safety evaluations for topical delivery systems.
Facing regulatory challenges? Discover the root causes of failed extractables and leachables (E&L) studies.
Conclusion: Mastering Extractables and Leachables Thresholds for Global Compliance
Achieving compliance with contemporary regulatory and compendial expectations requires the accurate and scientifically justified application of safety thresholds throughout the entire product lifecycle. By establishing appropriate analytical evaluation targets and aligning testing strategies with toxicological principles, manufacturers can effectively control chemical risks, satisfy global regulatory requirements, and protect patient safety.
Current regulatory expectations extend far beyond simple qualitative characterization. Regulatory agencies such as the FDA, EMA, and Health Canada increasingly expect sponsors to provide a comprehensive, evidence-based narrative linking material selection, extractables characterization studies, leachables assessments, and toxicological safety evaluations.
The proper implementation of SCT, QT, and AET concepts is essential for successful regulatory submissions. Correct application of these thresholds helps prevent patient exposure to potentially harmful contaminants, reduces unnecessary reporting of toxicologically insignificant compounds, and minimizes the likelihood of Complete Response Letters (CRLs), regulatory deficiencies, or post-approval manufacturing delays.
For organizations seeking support with ISO 10993-18 chemical characterization studies, statistically justified Uncertainty Factor derivations, and regulatory-compliant extractables and leachables strategies, consulting experienced specialists can help ensure both scientific rigor and global regulatory acceptance.
FAQs on Extractables and Leachables Thresholds
Extractables are compounds that can be pulled out of packaging, device, or manufacturing materials under aggressive lab conditions like strong solvents or elevated temperatures. They represent a worst-case chemical profile of the material. Leachables, on the other hand, are the subset of these compounds that actually migrate into the drug product during normal production, storage, or clinical use. Because of this, leachables are considered more directly relevant to patient exposure and real-world safety assessment.
The AET is derived by linking toxicological limits with measurable laboratory concentrations. It starts with the Safety Concern Threshold (SCT), which is divided by the Maximum Daily Dose (MDD) of the patient. This value is then adjusted using extraction study parameters such as solvent volume and contact ratio. In most cases, the final AET is further corrected using an Uncertainty Factor (UF) to reduce the risk of under-detection during instrumental analysis.
The May 2026 USP requirements significantly strengthen regulatory expectations for extractables and leachables control in pharmaceutical manufacturing. They make it essential for manufacturers to demonstrate that materials used in production systems do not release harmful chemical impurities above defined safety limits. Chapters such as USP <661.2> reinforce the need for systematic evaluation of polymeric and single-use components to ensure consistent product safety.
The TSL can only be used when the chemical structure of a compound is known and confirmed. Unidentified substances may belong to highly potent toxic groups that are not covered under general threshold assumptions. Without structural identification, there is no way to rule out high-risk categories such as mutagenic or carcinogenic substances. Therefore, unidentified compounds must undergo full analytical and toxicological evaluation before any threshold-based exclusion can be applied.
Cohort of concern compounds are a group of highly potent carcinogens that include substances like N-nitrosamines, PAHs, and aflatoxin-related chemicals. These compounds are so toxic that even extremely low exposures can pose significant health risks. Because of this, they are excluded from general thresholds like SCT or TSL. Instead, they require strict, compound-specific exposure limits based on detailed toxicological assessment.
An empirical RRF database helps laboratories understand how different chemicals respond during analytical testing compared to reference standards. Since detector responses vary between compounds, this database is used to measure and quantify those differences. The collected data is then used to calculate a statistically justified Uncertainty Factor (UF). This ensures more accurate interpretation of GC-MS and LC-MS results and reduces the chance of missing harmful compounds.
Route-to-route differences are corrected using bioavailability factors known as F6 under the draft ICH Q3E framework. These factors adjust toxicity data depending on how well a compound is absorbed through different exposure routes. For example, poorly absorbed compounds may require higher correction factors, while highly absorbed substances require lower adjustments. This ensures that systemic exposure is not underestimated when translating data from one administration route to another.
The Margin of Safety (MoS) is a key toxicological ratio used to evaluate chemical exposure from medical devices. It compares the tolerable intake level (TI) of a substance with the worst-case estimated exposure dose (EEDmax). If the MoS is equal to or greater than 1, it indicates that exposure remains within acceptable safety limits. This means the chemical is unlikely to cause harmful biological effects under expected clinical conditions.
Reference:
- Extractables and leachables in pharmaceutical products: Potential adverse effects and toxicological risk assessment. (2025). Pharmaceuticals. PubMed Central. https://pmc.ncbi.nlm.nih.gov/articles/PMC12846058/
- U.S. Food and Drug Administration. (2023, December). Quality considerations for topical ophthalmic drug products: Guidance for industry (Draft guidance, Revision 1). U.S. Department of Health and Human Services. https://www.fda.gov/media/183127/download
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). (2025). ICH Q3E guideline for extractables and leachables (Step 2 draft guideline). European Medicines Agency. https://www.ema.europa.eu/en/documents/scientific-guideline/draft-ich-q3e-guideline-extractables-leachables_en.pdf
- International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH). (2025, August 26). ICH Q3E: Guideline for extractables and leachables—Step 2 presentation. https://database.ich.org/sites/default/files/ICH_Q3E_Step2_Presentation_2025_0826.pdf

