
Introduction
Implementing the ICH Q6B Guidelines for Biological Characterisation requires a comprehensive, multi-orthogonal analytical strategy to verify that a proposed biosimilar aligns with its reference product at the molecular level, ensuring that no clinically meaningful differences are present. Since biopharmaceuticals are manufactured using living cellular systems, such as Chinese Hamster Ovary (CHO) cells, they naturally exhibit inherent biological variability, making exact duplication unattainable. Consequently, developers must establish that the biosimilar is highly similar to the reference biologic by rigorously following the ICH Q6B Guidelines for Biological Characterisation, which serve as the internationally recognized framework for evaluating physicochemical characteristics, structural integrity, and biological function. By incorporating advanced mass spectrometry techniques and higher-order structural analyses early in the product development process, organizations can accurately define the molecule’s critical quality attributes (CQAs), thereby minimizing residual uncertainty and reducing the need for extensive clinical studies.
Related Resource: Learn more about defining Critical Quality Attributes (CQAs) in Biosimilars to guide your early-stage development.
Article Summary:
- ICH Q6B serves as the global framework for biosimilar characterization, requiring developers to demonstrate high analytical similarity to the reference biologic through comprehensive molecular, structural, and functional evaluation rather than proving identical composition.
- A multi-orthogonal analytical strategy is essential because no single technique can fully characterize complex biologics. Combining complementary technologies provides a complete understanding of critical quality attributes (CQAs) while strengthening regulatory confidence.
- Primary structure and higher-order structure must be thoroughly verified to confirm the correct amino acid sequence, terminal modifications, protein folding, conformational stability, and overall structural integrity that directly influence therapeutic performance.
- Post-translational modifications (PTMs), especially glycosylation and disulfide bonding, require detailed characterization since these molecular features significantly affect biological activity, pharmacokinetics, immunogenicity, and long-term product consistency.
- Purity assessment extends beyond a single purity value by identifying and quantifying both product-related variants, such as aggregates and charge variants, and process-related impurities, including host cell proteins, residual DNA, and manufacturing contaminants.
- The totality of the evidence approach relies on extensive analytical comparability across multiple production batches, allowing regulators to minimize uncertainty, reduce the dependence on large clinical studies, and support extrapolation to additional therapeutic indications.
- Advanced mass spectrometry and specialized analytical expertise play a central role in successful biosimilar development, enabling developers to generate robust regulatory data, accelerate approvals, and maintain consistent product quality throughout the development lifecycle.
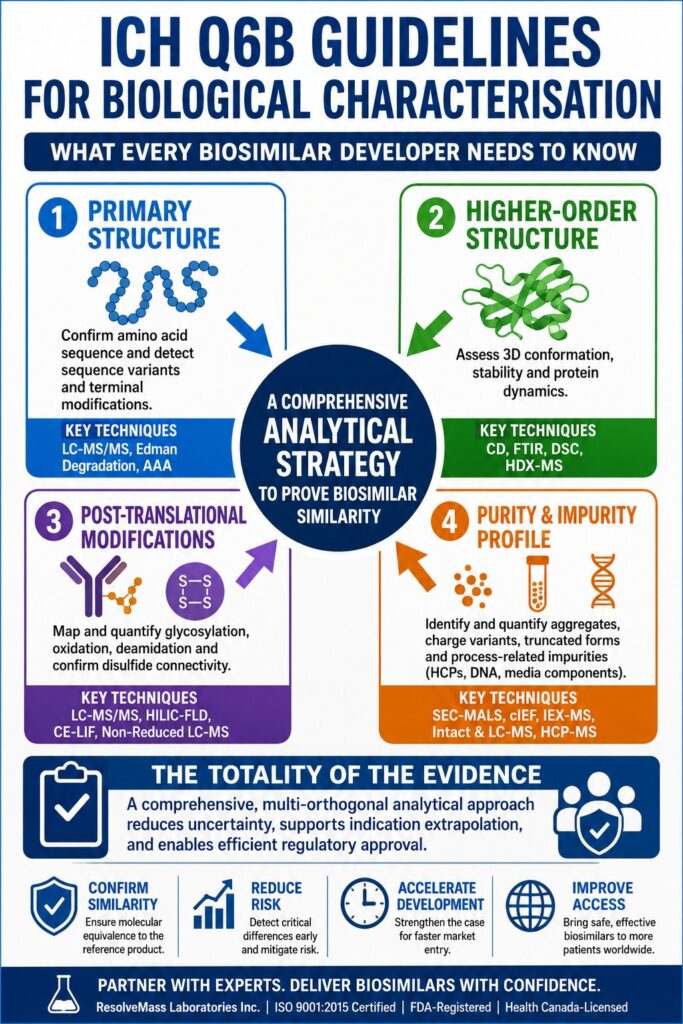
Strategic Execution of the ICH Q6B Guidelines for Biological Characterisation
The successful implementation of the ICH Q6B Guidelines for Biological Characterisation requires a robust analytical package capable of establishing a detailed molecular fingerprint, which forms the cornerstone of the regulatory totality of the evidence approach. Regulatory authorities, including the FDA and EMA, do not regard biosimilar assessment as a routine quality control procedure. Instead, they evaluate it as a rigorous, science-driven, and iterative comparability exercise that encompasses extensive analysis across numerous manufacturing batches.
Related Resource: Discover how to structure a rigorous comparability exercise in biosimilar development to meet international regulatory standards.
To satisfy these regulatory requirements, developers must evaluate a predefined range of structural parameters using multiple orthogonal analytical techniques. Employing several complementary methods ensures that the limitations or potential bias associated with one analytical platform are offset by another, resulting in a more reliable and comprehensive characterization of the biological product.
| Structural Tier | Analytical Objective | Primary Technologies Deployed |
|---|---|---|
| Primary Structure | Confirm the precise amino acid sequence, terminal residues, and sequence variants. | LC-MS/MS Peptide Mapping, Edman Degradation, Amino Acid Analysis (AAA) |
| Higher-Order Structure (HOS) | Evaluate alpha-helices, beta-sheets, protein folding, and conformational stability. | Circular Dichroism (CD), Fourier-Transform Infrared (FTIR), Hydrogen-Deuterium Exchange MS (HDX-MS) |
| Post-Translational Modifications | Quantify glycosylation patterns, deamidation, oxidation, and glycation levels. | High-Resolution Mass Spectrometry (HRMS), HILIC-FLD, Site-Specific Glycopeptide Mapping |
| Purity and Impurities | Detect aggregates, truncated variants, host cell proteins (HCPs), and residual DNA. | Size Exclusion Chromatography (SEC-MALS), Capillary Electrophoresis (CE-SDS), HCP-MS |
Organizations such as ResolveMass Laboratories Inc., an ISO 9001:2015 certified and FDA-registered Contract Research Organization (CRO), offer the sophisticated mass spectrometry capabilities necessary to conduct complex comparability studies in accordance with the ICH Q6B Guidelines for Biological Characterisation. Through the use of advanced analytical instrumentation, researchers can move beyond conventional qualitative characterization and achieve precise quantitative evaluation using Multi-Attribute Monitoring (MAM).
Explore our specialized biosimilar characterization services to discover how we can support your analytical testing goals.
Primary Structural Analysis and Sequence Integrity
Primary structural analysis verifies the exact linear amino acid sequence of a biologic while identifying unintended sequence variants and terminal modifications that may influence the molecule’s functional architecture. According to the ICH Q6B Guidelines for Biological Characterisation, demonstrating a complete primary sequence match between the biosimilar and its reference product is an essential regulatory expectation.
Peptide mapping performed using liquid chromatography-tandem mass spectrometry (LC-MS/MS) remains the gold-standard analytical approach for meeting this requirement. During this process, the protein undergoes controlled enzymatic digestion, commonly using orthogonal proteases such as trypsin, chymotrypsin, or Endoproteinase Glu-C, to generate overlapping peptide fragments. These fragments are subsequently analyzed using high-resolution quadrupole time-of-flight (Q-TOF) or Orbitrap mass spectrometers, enabling accurate sequence determination through precise fragment ion mass measurements.
In addition to internal sequence verification, terminal modifications require thorough assessment. N-terminal pyroglutamination and C-terminal lysine clipping represent common microheterogeneities capable of altering the charge profile of monoclonal antibodies (mAbs). Although C-terminal lysine clipping generally exerts minimal influence on biological activity, deamidation occurring within the complementarity-determining regions (CDRs) can substantially reduce target-binding affinity. When terminal residues cannot be confidently identified through mass spectrometry because of blockage or ambiguity, Edman degradation (gas-phase sequencing) is employed to sequentially identify amino acids based on chromatographic elution patterns.
Additionally, the precise determination of overall amino acid composition is achieved through acid hydrolysis followed by chromatographic separation. This analysis is essential for calculating the protein’s extinction coefficient, a critical parameter used to determine the exact concentration of the active pharmaceutical ingredient according to the Beer-Lambert Law.
Higher-Order Structure (HOS) Comparability
Higher-Order Structure (HOS) comparability ensures that the intricate three-dimensional architecture of the biosimilar, encompassing its secondary, tertiary, and quaternary structures, closely mirrors that of the reference biologic. The biological function and immunogenic profile of a protein are fundamentally dependent on its native three-dimensional conformation. Improperly folded proteins often expose previously hidden epitopes, which can stimulate undesirable anti-drug antibody (ADA) responses in patients.
To evaluate HOS, developers employ an integrated combination of spectroscopic and thermodynamic analytical techniques. Circular Dichroism (CD) and Fourier-transform infrared (FTIR) spectroscopy are routinely used to characterize secondary structural elements by accurately measuring the relative proportions of alpha-helices and beta-sheets. Differential scanning calorimetry (DSC) is applied to determine thermal stability and protein unfolding transition temperatures, providing valuable insight into the overall thermodynamic stability of the formulated biologic.
Whenever subtle spectral variations are observed, Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) offers highly detailed, residue-level information regarding protein dynamics and conformational similarity. By monitoring the rate at which deuterium replaces hydrogen atoms along the protein backbone in solution, HDX-MS identifies solvent-accessible regions throughout the molecule. This detailed mapping confirms that critical antigen-binding domains and Fc regions maintain structural integrity and exhibit conformational equivalence with the reference product.
Navigating Post-Translational Modifications (PTMs)
Navigating Post-Translational Modifications (PTMs) requires comprehensive mapping of the enzyme-mediated chemical modifications that occur following protein translation, as these molecular alterations directly influence therapeutic safety, pharmacokinetic clearance, and effector function. The ICH Q6B Guidelines for Biological Characterisation require developers to thoroughly quantify PTM heterogeneity while establishing well-defined acceptance criteria for each critical modification.
Read our comprehensive guide on mapping post-translational modifications (PTMs) in biosimilars.
The Criticality of Glycosylation Profiling
For monoclonal antibodies, glycosylation at the Asn297 residue within the Fc region represents a Critical Quality Attribute (CQA) because it directly regulates antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). The glycoform distribution is particularly sensitive to variations in biomanufacturing processes, making glycosylation profiling one of the most critical aspects of biosimilar development.
In particular, the extent of core fucosylation requires careful monitoring because afucosylated antibodies display significantly greater affinity for the FcγRIIIa activating receptor expressed on immune effector cells, resulting in enhanced ADCC activity. Conversely, the presence of non-human glycan epitopes, including N-glycolylneuraminic acid (Neu5Gc) and alpha-1,3-galactose, which may originate from murine expression systems, can substantially increase the risk of patient immunogenicity.
To achieve comprehensive glycosylation characterization, advanced analytical platforms such as HILIC-FLD (Hydrophilic Interaction Liquid Chromatography with Fluorescence Detection) and Capillary Electrophoresis with Laser-Induced Fluorescence (CE-LIF) are routinely used alongside LC-MS/MS glycopeptide mapping. This multi-orthogonal analytical strategy enables developers to evaluate released glycans for overall population distribution while simultaneously using site-specific glycopeptide mapping to verify accurate glycosylation site occupancy and structural consistency.
Dive deeper into the advanced mass spectrometry methodologies used for glycosylation analysis of biosimilars.
Disulfide Bond Connectivity and Scrambling
The formation of correct disulfide bonds is critical for preserving both the structural integrity and biological functionality of multi-chain biologics. Under the ICH Q6B Guidelines for Biological Characterisation, developers must verify the precise positions of both intra-chain and inter-chain disulfide bridges. Any occurrence of disulfide bond scrambling, incomplete bond formation, or the presence of free sulfhydryl groups can disrupt proper protein folding and significantly increase the likelihood of protein aggregation. To evaluate these structural attributes, non-reduced peptide mapping workflows combined with LC-MS are employed to identify native disulfide linkages, while free thiol assays are used to quantify unpaired cysteine residues. Together, these complementary analytical approaches provide a comprehensive assessment of the molecule’s structural integrity and long-term stability.
Defining Purity and Establishing Impurity Profiles
Defining purity and establishing impurity profiles in accordance with the ICH Q6B Guidelines for Biological Characterisation requires the careful identification, differentiation, and quantification of both product-related structural variants and process-related contaminants. Owing to the inherent complexity of biologics, purity cannot be represented by a single numerical value. Instead, it is defined through a comprehensive impurity profile that characterizes acceptable levels of molecular heterogeneity while ensuring consistent product quality and safety.
Product-Related Impurities
Product-related impurities consist of molecular variants of the intended biologic that are generated during protein expression, purification, formulation, or long-term storage. These variants generally exhibit reduced or absent biological activity and must be thoroughly characterized.
High-Molecular-Weight (HMW) Aggregates: Protein aggregation represents one of the most significant immunogenic risks associated with biologic therapeutics. Size Exclusion Chromatography (SEC) coupled with triple detection using Multi-Angle Light Scattering (MALS), UV, and Refractive Index (RI) detection remains the established analytical standard for accurately quantifying dimers and higher-order protein aggregates.
Understand how to mitigate immunogenic risks with aggregation analysis in biosimilars.
Charge Variants: Chemical modifications such as deamidation, oxidation, and glycation generate acidic and basic charge variants within the protein population. These heterogeneities are characterized using Capillary Isoelectric Focusing (cIEF) together with Ion-Exchange Chromatography coupled with Mass Spectrometry (IEX-MS), allowing detailed isolation and molecular characterization of individual charge species.
Learn how high-resolution mass spectrometry enhances charge variant analysis in biosimilars.
Truncated Forms: Proteolytic processing that occurs during manufacturing can produce truncated protein variants or clipped species. These structural variants are evaluated through intact mass analysis in combination with LC-MS, enabling precise identification and quantification of cleavage products.
Process-Related Impurities and the Shift to HCP-MS
Process-related impurities include trace residual materials originating from the host expression system, such as Chinese Hamster Ovary (CHO) cells, along with components derived from culture media and downstream purification processes. These impurities include Host Cell Proteins (HCPs), host cell DNA, antibiotics such as Kanamycin, inducers including IPTG, and formulation-related surfactants such as Tween 20 and Poloxamer 188.
Traditionally, Host Cell Proteins (HCPs) were monitored almost exclusively through Enzyme-Linked Immunosorbent Assays (ELISA). However, conventional ELISA methodologies depend on polyclonal antibodies that may not detect low-abundance proteins, weakly immunogenic impurities, or highly active enzymatic contaminants. For instance, trace levels of hamster phospholipase B-like 2 (PLBL2) or specific lipases may co-purify with the drug substance and subsequently degrade polysorbate excipients over time, potentially leading to severe formulation instability and product failure.
Read more about advanced impurity profiling of biosimilars to ensure robust product purity and compliance.
To address these analytical limitations, regulatory agencies are increasingly encouraging developers to adopt Host Cell Protein Mass Spectrometry (HCP-MS) as part of their impurity characterization strategy. Laboratories utilizing native digestion LC-MS workflows can identify and accurately quantify individual trace HCPs at sub-ppm concentrations without depending on specialized antibody reagents. This targeted proteomics approach provides detailed molecular insight that supports data-driven optimization of downstream purification processes while enhancing patient safety and preserving polysorbate stability. ResolveMass Laboratories Inc. operates advanced mass spectrometry platforms specifically optimized for detecting and quantifying these difficult-to-identify process-related impurities within highly complex biological matrices.
Learn how leveraging a targeted proteomics approach for biosimilars overcomes traditional ELISA limitations.
The Totality of the Evidence and Biosimilar Extrapolation
The totality of the evidence represents the fundamental regulatory principle governing biosimilar approval, relying on a comprehensive analytical data package to establish a detailed molecular fingerprint while substantially reducing the need for extensive comparative clinical trials. Regulatory authorities, including the FDA and EMA, evaluate biosimilarity using a hierarchical framework in which highly sensitive physicochemical characterization and functional assessment form the broad scientific foundation supporting subsequent nonclinical and clinical evidence.
Conducting a biosimilar comparability study generally involves evaluating approximately six to ten independent batches of the proposed biosimilar alongside an equivalent number of batches from the innovator reference product. Following the ICH Q6B Guidelines for Biological Characterisation, developers establish scientifically justified acceptance ranges for every Critical Quality Attribute (CQA). When comprehensive analytical results consistently demonstrate that the biosimilar falls within the natural variability observed for the reference product across all orthogonal analytical methods, the remaining uncertainty regarding biosimilarity is significantly reduced.

Access our insights on designing and executing compliant biosimilar comparability studies.
This extensive analytical evidence forms the regulatory basis for the legal principle of extrapolation of indications. Extrapolation allows a biosimilar to receive approval for multiple therapeutic indications already authorized for the reference biologic, such as both rheumatoid arthritis and plaque psoriasis for a TNF-alpha inhibitor, without requiring separate Phase III clinical trials for every individual patient population. Consequently, establishing a strong analytical foundation extends beyond regulatory compliance; it also serves as a major contributor to commercial success by accelerating market entry while substantially reducing overall clinical development costs.
Conclusion
Achieving compliance with the ICH Q6B Guidelines for Biological Characterisation is an essential requirement for successful biosimilar development, establishing the scientific rigor necessary to demonstrate structural equivalence, functional consistency, and clinical safety. From utilizing high-resolution LC-MS/MS for primary sequence confirmation and detailed glycosylation profiling to applying HDX-MS for comprehensive higher-order structure verification, developers must generate an exceptionally robust and scientifically defensible analytical data package. By thoroughly characterizing process-related impurities through HCP-MS and ensuring that product-related heterogeneities closely align with the molecular fingerprint of the reference biologic, organizations can leverage the totality of the evidence to facilitate efficient regulatory approval and support indication extrapolation.
Executing this advanced level of Multi-Attribute Monitoring (MAM) requires collaboration with a highly specialized analytical partner possessing extensive expertise in biologic characterization. ResolveMass Laboratories Inc., an ISO 9001:2015 certified, FDA-registered, and Health Canada-licensed facility, offers the scientific knowledge and state-of-the-art mass spectrometry technologies required to navigate complex regulatory expectations and support the successful commercialization of critical biologic therapies in global markets.
To learn how our advanced mass spectrometry capabilities can strengthen your biosimilar characterization strategy and support seamless regulatory compliance, visit our Contact Us page.
Frequently Asked Questions (FAQs)
LC-MS/MS peptide mapping is widely recognized as the benchmark technique for primary structural characterization because it provides highly accurate verification of the protein’s amino acid sequence. The protein is enzymatically digested into smaller peptides, which are then analyzed using high-resolution mass spectrometry. This approach confirms sequence integrity while simultaneously detecting sequence variants and identifying important post-translational modifications with exceptional precision.
Multi-Attribute Monitoring (MAM) is an advanced LC-MS-based analytical strategy that enables multiple Critical Quality Attributes (CQAs) to be evaluated within a single workflow. It allows simultaneous monitoring of attributes such as glycosylation, oxidation, deamidation, and other molecular modifications. This comprehensive analysis improves manufacturing consistency, supports batch-to-batch comparability, and provides the detailed analytical evidence expected by regulatory agencies.
Glycosylation plays a significant role in determining the biological activity of monoclonal antibody biosimilars, particularly through modifications at the Asn297 residue in the Fc region. Even subtle differences in glycan composition can affect interactions with immune cell receptors, influencing mechanisms such as antibody-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC). Therefore, maintaining a glycosylation profile that closely matches the reference product is essential for consistent therapeutic performance.
Host Cell Proteins (HCPs) are residual impurities originating from the cells used to manufacture biologic products. If not adequately removed, these proteins may contribute to immunogenic responses or interfere with formulation stability by degrading excipients such as polysorbates. While ELISA has traditionally been used for HCP detection, modern analytical strategies increasingly utilize HCP-MS, which offers greater sensitivity and enables identification and quantification of trace-level impurities.
Higher-Order Structure (HOS) comparability confirms that a biosimilar possesses the same three-dimensional conformation as its reference product. Analytical techniques such as Circular Dichroism (CD), Fourier-Transform Infrared (FTIR) spectroscopy, and Hydrogen-Deuterium Exchange Mass Spectrometry (HDX-MS) are commonly used to assess structural similarity. Proper protein folding minimizes the exposure of unwanted epitopes and reduces the likelihood of anti-drug antibody (ADA) formation and other immunogenic responses.
The totality of the evidence approach places significant emphasis on comprehensive analytical, structural, and functional characterization before extensive clinical testing is considered. When detailed analytical studies demonstrate a high degree of similarity between a biosimilar and its reference product, regulatory authorities may accept a reduced clinical development program. This scientific approach can shorten development timelines, support indication extrapolation, and substantially lower overall development costs.
Disulfide bond scrambling occurs when cysteine residues form incorrect disulfide linkages or remain as unpaired sulfhydryl groups instead of creating their intended structural bonds. Such abnormalities can alter the protein’s native conformation, compromise its biological function, and increase its susceptibility to degradation and aggregation. As a result, maintaining correct disulfide bond connectivity is essential for preserving product stability, efficacy, and patient safety.
An orthogonal analytical strategy involves evaluating the same quality attribute using multiple independent analytical techniques, each based on different scientific principles. Combining methods such as mass spectrometry, spectroscopy, chromatography, and electrophoresis provides a more complete understanding of the molecule while minimizing the limitations of any individual technique. This comprehensive approach strengthens the reliability of comparability assessments and increases confidence in regulatory submissions.
Developers establish acceptable variability by analyzing multiple commercial batches of the reference biologic to understand its natural manufacturing variability over time. The collected analytical data are used to define statistically justified acceptance ranges for each Critical Quality Attribute (CQA). A proposed biosimilar must consistently fall within these predefined limits to demonstrate high similarity and satisfy regulatory expectations for approval.
Reference:
- U.S. Food and Drug Administration. (2015, April). Quality considerations in demonstrating biosimilarity of a therapeutic protein product to a reference product: Guidance for industry. https://www.fda.gov/media/135612/download
- European Medicines Agency (EMA). ICH Q6B: Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products – Scientific Guideline. Available at: https://www.ema.europa.eu/en/ich-q6b-specifications-test-procedures-acceptance-criteria-biotechnological-biological-products-scientific-guideline
- Berkowitz S, Engen JR, Mazzeo JR, Jones GB. Analytical Tools for Characterizing Biopharmaceuticals and the Implications for Biosimilars. Nature Reviews Drug Discovery. 2012;11(7):527–540. doi:10.1038/nrd3746. Available at: https://pmc.ncbi.nlm.nih.gov/articles/PMC3714370/. Accessed July 8, 2026.
- International Council for Harmonisation (ICH). Session III: ICH Q6B – Specifications: Test Procedures and Acceptance Criteria for Biotechnological/Biological Products [Training Presentation]. Available at: https://admin.ich.org/sites/default/files/inline-files/SESSION_III_ICHQ6B_Specifications.pdf. Accessed July 8, 2026. _

