
Introduction:
The role of PK/PD studies in biosimilar development has become one of the most closely watched questions in biologics regulatory strategy, as sponsors look for scientifically sound ways to bring cost-effective biosimilars to patients faster without compromising safety or efficacy. Rather than repeating the large, resource-intensive clinical trials used to approve the original reference biologic, biosimilar developers rely on a stepwise body of evidence — starting with analytical characterization and culminating, when necessary, in clinical studies. Pharmacokinetic and pharmacodynamic (PK/PD) data sit at the center of this totality-of-evidence approach, offering a sensitive, human-relevant way to confirm that a proposed biosimilar behaves the same way in the body as its reference product. Understanding how FDA and EMA regulatory pathways treat this evidence — and how biosimilars differ from small-molecule generics in the first place, given the well-documented biosimilar vs. generic drug differences — is essential context before diving into what PK/PD studies actually measure and whether they can stand in for large-scale clinical efficacy trials.
Summary:
- Pharmacokinetic/pharmacodynamic (PK/PD) studies compare how a proposed biosimilar and its reference product are absorbed, distributed, metabolized, and eliminated, and how they act on the body’s biological targets.
- Regulatory agencies (FDA, EMA, Health Canada) use PK/PD equivalence as a foundational pillar of the biosimilar “totality-of-evidence” approach, alongside analytical, nonclinical, and clinical immunogenicity data.
- In many cases, robust PK/PD similarity — combined with strong analytical and functional characterization — can reduce or even eliminate the need for large, comparative clinical efficacy trials.
- PK/PD studies cannot fully replace clinical trials when a biosimilar’s mechanism of action is complex, the target patient population is heterogeneous, or immunogenicity risk is high.
- ResolveMass Laboratories supports biosimilar developers with the analytical and bioanalytical foundation — including higher-order structure characterization, potency assays, and impurity profiling — that underpins credible PK/PD and comparability packages.
1: Role of PK/PD Studies in Biosimilar Development: Why They Matter
The role of PK/PD studies in biosimilar development is to demonstrate that a proposed biosimilar behaves the same way in the human body as its reference product, without requiring a full independent efficacy program built from scratch. Instead of proving a new drug works, biosimilar sponsors must prove their molecule is highly similar to an already-approved product through rigorous biosimilar comparability studies — and PK/PD data is often the most direct, sensitive way to show that similarity in a living system.
Because biosimilars are large, structurally complex molecules (monoclonal antibodies, fusion proteins, biosimilar insulins, and other biologics), even minor differences in glycosylation, aggregation, or higher-order structure can influence how the molecule is absorbed, cleared, or how it engages its target receptor. This is why sponsors invest heavily in biosimilar characterization using mass spectrometry and rigorous evaluation of critical quality attributes (CQAs) long before a PK/PD study is ever run. PK/PD studies act as a sensitive bridge between analytical characterization in the lab and clinical outcomes in patients.
2: What Do PK/PD Studies in Biosimilar Development Actually Measure?
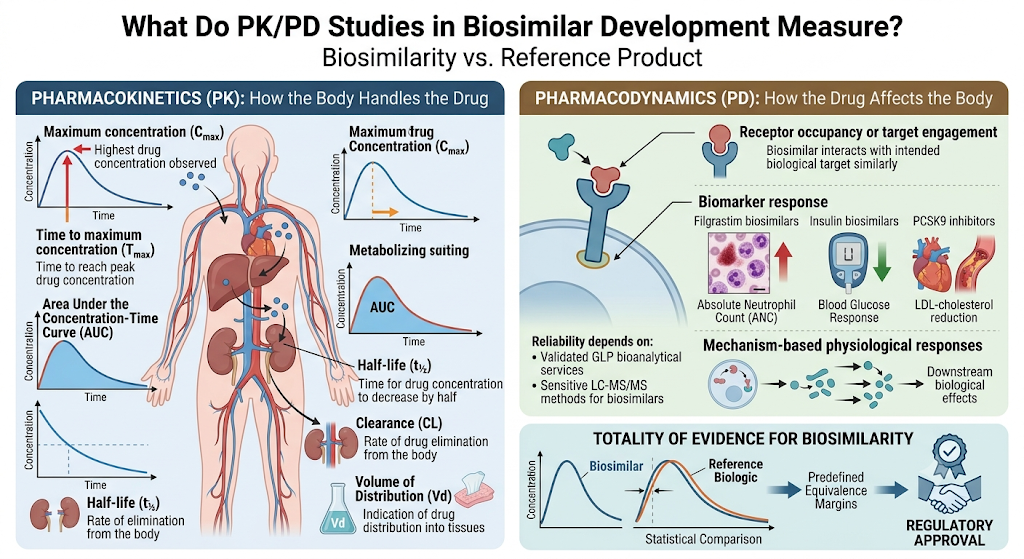
PK/PD studies measure two connected but distinct things: how the body handles the drug (pharmacokinetics) and how the drug affects the body (pharmacodynamics). Generating this data reliably depends on validated GLP bioanalytical services and sensitive LC-MS/MS methods for biosimilars capable of detecting small concentration differences between test and reference products.
Pharmacokinetic (PK) parameters typically assessed include:
Pharmacokinetic studies evaluate the drug’s absorption, distribution, metabolism, and elimination by measuring parameters such as:
- Maximum concentration (Cmax): The highest drug concentration observed in the bloodstream after administration.
- Time to maximum concentration (Tmax): The time required to reach peak drug concentration.
- Area Under the Concentration-Time Curve (AUC): A measure of the body’s overall exposure to the drug over time.
- Half-life (t½): The time required for the drug concentration to decrease by half, reflecting how long it remains in the body.
- Clearance (CL): The rate at which the drug is eliminated from the body.
- Volume of Distribution (Vd): An indicator of how extensively the drug distributes into body tissues.
Pharmacodynamic (PD) markers typically assessed include:
Pharmacodynamic studies assess the biological effects of the drug by measuring relevant biomarkers and functional responses, including:
- Receptor occupancy or target engagement: Demonstrates whether the biosimilar interacts with its intended biological target similarly to the reference product.
- Biomarker response (e.g., absolute neutrophil count for filgrastim biosimilars, glucose response for insulin biosimilar characterization, or LDL-cholesterol reduction for PCSK9 inhibitors)
- Biomarker response: Examples include absolute neutrophil count (ANC) for filgrastim biosimilars, blood glucose response for insulin biosimilars, and LDL-cholesterol reduction for PCSK9 inhibitor biosimilars.
- Mechanism-based physiological responses: Evaluates downstream biological effects directly associated with the drug’s mechanism of action.
Together, these datasets generate a PK/PD profile that regulators compare statistically against the reference product, an exercise that relies heavily on a well-designed biosimilar bioanalysis strategy and predefined equivalence margins.
The combined PK and PD data generate a comprehensive PK/PD profile, which regulators compare statistically with the reference biologic using predefined equivalence margins. Supported by robust bioanalytical testing and scientifically sound study design, these data form a key component of the totality of evidence used to demonstrate biosimilarity and support regulatory approval.
3: Where Do PK/PD Studies Sit in the Regulatory Pathway?
PK/PD studies sit early in the clinical phase of biosimilar development, typically as the first human comparative study following analytical and nonclinical characterization. This sequencing reflects the “stepwise” or totality-of-evidence model used by FDA, EMA, and Health Canada, and it starts well before clinical dosing, often with cell line development for biosimilars and extensive structural analysis.
| Development Stage | Purpose | Typical Study Type |
|---|---|---|
| Analytical similarity | Establish structural and functional comparability | Peptide mapping, disulfide bond mapping, charge variant analysis, and higher-order structure characterization |
| Nonclinical | Confirm biological activity and safety signals | In vitro binding/potency assays, animal PK/tox studies, forced degradation of biosimilars |
| Clinical PK/PD | Confirm human PK/PD equivalence | Single-dose, crossover PK/PD study in healthy volunteers or patients |
| Clinical efficacy/safety (if required) | Confirm no clinically meaningful differences remain | Comparative efficacy trial with immunogenicity monitoring |
A biosimilar only advances to a large comparative efficacy trial if residual scientific uncertainty remains after the analytical, nonclinical, and PK/PD steps, and after sponsors have completed the full slate of analytical tests required for biosimilar regulatory submission.
4: Can PK/PD Studies Replace Clinical Efficacy Trials?
Yes, in many biosimilar programs, strong PK/PD similarity combined with robust analytical characterization can reduce, streamline, or in some cases eliminate the need for a large comparative efficacy trial. This is one of the central efficiencies the biosimilar pathway was designed to create, since repeating full Phase III efficacy programs for molecules that are already highly similar to an approved reference product offers limited additional scientific value.
Regulatory guidance from both FDA and EMA acknowledges that when a biosimilar’s mechanism of action is well understood, its PD markers are validated and sensitive to differences, and analytical similarity is high — as confirmed through methods like native mass spectrometry for biosimilars and intact mass analysis — a comparative efficacy trial may add little incremental information. Several approved biosimilars have relied primarily on PK/PD equivalence data rather than a large-scale efficacy trial.
PK/PD data is more likely to support waiving a clinical efficacy trial when:
- The mechanism of action is simple and well-characterized (e.g., replacement therapies, receptor agonists/antagonists)
- Validated, clinically relevant PD biomarkers exist, often identified through a targeted proteomics approach for biosimilars
- The reference product’s clinical pharmacology is well established
- Analytical similarity data shows a high degree of structural and functional match, with no gaps that could trigger a biosimilar comparability failure
A clinical efficacy trial is still generally required when:
- The molecule has multiple mechanisms of action or complex immune-mediated effects
- PD biomarkers are not sufficiently sensitive or validated
- The patient population is heterogeneous, making PD response harder to interpret
- Immunogenicity risk is significant and could affect long-term efficacy or safety
Sponsors who underestimate these risk factors are frequently the same ones who end up studying why biosimilars fail regulatory approval after the fact, rather than designing around them from the start.
5: Why Can’t PK/PD Studies Fully Replace Clinical Trials in Every Case?
PK/PD studies cannot fully replace clinical trials in every case because they measure surrogate markers of drug behavior, not always the final clinical outcome patients and physicians care about. For molecules where PD markers do not reliably predict efficacy, or where subtle immunogenic responses could only emerge after repeated dosing in a real patient population, regulators still require confirmatory clinical data.
Immunogenicity is a particularly important limitation. Anti-drug antibodies can develop gradually and may not be detectable in a short-duration PK/PD study, which is why a thorough immunogenicity assessment in biosimilar development, supported by dedicated anti-drug antibody (ADA) assay development, is an essential complement to PK/PD equivalence data.
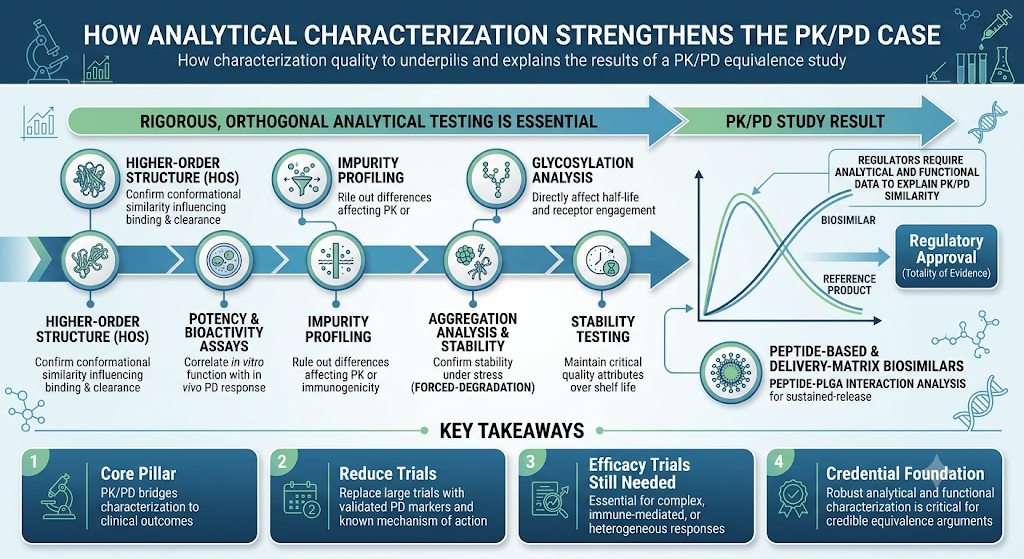
6: How Analytical Characterization Strengthens the PK/PD Case
The strength of a PK/PD equivalence argument depends heavily on the quality of the analytical and functional data supporting it. Regulators expect sponsors to demonstrate that any observed PK/PD similarity is consistent with, and explained by, the molecule’s structural and functional profile — a case that is only as strong as the peptide mapping and sequence analysis and peptide biosimilar characterization by LC-MS behind it.
This is where rigorous, orthogonal analytical testing becomes essential to a biosimilar’s overall comparability package:
- Higher-order structure (HOS) characterization to confirm conformational similarity that can influence receptor binding and clearance
- Potency and bioactivity assays to correlate in vitro function with in vivo PD response
- Impurity profiling of biosimilars to rule out product-related differences that could affect PK behavior or immunogenicity
- Glycosylation analysis of biosimilars, since these post-translational modifications directly affect half-life and receptor engagement
- Aggregation analysis and forced-degradation testing to confirm the molecule remains stable and comparable under stress conditions, an approach detailed further in our dedicated biosimilar forced degradation study resource
- Biosimilar stability testing to confirm the product maintains its critical quality attributes over its intended shelf life
For peptide-based and delivery-matrix biosimilars, additional considerations such as peptide-PLGA interaction analysis may also factor into how closely PK/PD behavior tracks with the reference product, particularly for sustained-release formulations. These pressures echo the broader generic injectable market trends pushing sponsors toward faster, more defensible comparability packages.
A well-designed analytical package doesn’t just support the PK/PD study — it helps explain why the PK/PD results look the way they do, which is exactly what regulators are looking for in a totality-of-evidence submission.
Key Takeaways
- PK/PD studies are a core pillar of biosimilar development, bridging analytical characterization and clinical outcomes.
- They can reduce or replace the need for large comparative efficacy trials when the mechanism of action is well understood and PD markers are validated.
- Clinical efficacy trials remain necessary for complex, immune-mediated, or heterogeneous-response biologics.
- Robust analytical and functional characterization is the foundation that makes a PK/PD equivalence argument credible to regulators.
Conclusion:
The role of PK/PD studies in biosimilar development is best understood as the scientific bridge between laboratory-based analytical similarity and real-world clinical performance. When a biosimilar’s mechanism of action is well understood and its PD markers are validated and sensitive, robust PK/PD equivalence data can meaningfully reduce, or in some cases replace, the need for a large comparative efficacy trial — reflecting the efficiency the biosimilar pathway was built to deliver. At the same time, PK/PD studies are not a universal substitute for clinical trials: complex mechanisms, heterogeneous patient populations, and immunogenicity risk can all mean confirmatory clinical data is still required. In every case, the credibility of a PK/PD equivalence argument rests on the strength of the analytical and functional characterization behind it, making rigorous, well-designed comparability testing a non-negotiable foundation of any biosimilar development program.
Frequently Asked Questions:
PK/PD studies have become increasingly important because they provide highly sensitive and objective evidence of biosimilarity. Unlike clinical efficacy trials, they directly compare drug exposure and biological activity using measurable endpoints. Advances in analytical technologies have made these studies more reliable for detecting subtle differences between a biosimilar and its reference product. Regulatory agencies now recognize that robust PK/PD data can often answer critical scientific questions more efficiently than large clinical trials. This approach reduces unnecessary patient exposure to comparative efficacy studies while maintaining rigorous standards for quality, safety, and effectiveness.
PK/PD studies generally require fewer participants and shorter study durations than traditional clinical efficacy trials. Because they use quantitative endpoints, they can detect biosimilarity with greater statistical sensitivity and lower variability. This streamlined approach reduces clinical development costs, minimizes operational complexity, and accelerates regulatory submissions. Faster development timelines also help manufacturers bring affordable biosimilars to market sooner. Ultimately, this benefits healthcare systems by improving patient access to high-quality biologic therapies at lower costs.
Biologics with well-characterized mechanisms of action and validated pharmacodynamic biomarkers are often ideal candidates for PK/PD-based development. Examples include products such as insulin and filgrastim, where measurable biological responses closely reflect clinical activity. When strong analytical similarity and sensitive PK/PD endpoints are available, regulators may reduce or waive comparative clinical efficacy trials. However, each biosimilar is evaluated individually based on its molecular complexity, available biomarkers, and scientific evidence. A risk-based approach is always applied during regulatory review.
Regulatory agencies assess several scientific factors before deciding if a comparative clinical efficacy trial is necessary. These include the availability of validated PD biomarkers, analytical similarity results, pharmacokinetic comparability, mechanism of action, and immunogenicity risk. If these studies provide sufficient evidence to demonstrate biosimilarity, additional efficacy trials may not be required. However, products with complex mechanisms or unresolved scientific uncertainties may still need comparative clinical data. The decision is made on a case-by-case basis using the totality of evidence.
Validated biomarkers provide measurable indicators of a drug’s biological activity and are essential for demonstrating pharmacodynamic similarity. They allow researchers to compare how the biosimilar and reference product affect the body under controlled conditions. Reliable biomarkers increase the sensitivity of PK/PD studies by detecting even minor differences in biological response. This improves confidence in the study results and supports regulatory decision-making. When appropriate biomarkers are available, they can significantly reduce the need for large comparative efficacy trials.
Immunogenicity assessment evaluates whether a biologic triggers an unwanted immune response that could affect its safety or effectiveness. Anti-drug antibodies may alter pharmacokinetics, reduce therapeutic activity, or increase adverse reactions. Therefore, immunogenicity testing complements PK/PD studies by providing additional evidence about the biosimilar’s clinical performance. Regulatory agencies consider immunogenicity an essential component of the totality of evidence. Together, these assessments help ensure that the biosimilar performs similarly to the reference product in patients.
The totality of evidence approach combines data from analytical characterization, functional assays, PK/PD studies, immunogenicity assessments, and, where necessary, clinical studies. Rather than relying on a single type of evidence, regulators evaluate all available scientific information collectively. This comprehensive assessment provides greater confidence that the proposed biosimilar has no clinically meaningful differences from the reference biologic. The approach enables a more efficient and scientifically robust evaluation process while maintaining strict standards for product quality, safety, and efficacy.
Advanced bioanalytical testing ensures that drug concentrations and pharmacodynamic responses are measured accurately and consistently throughout a study. Techniques such as LC-MS/MS, ligand-binding assays, and biomarker analysis provide highly sensitive and reproducible results. Validated analytical methods minimize variability and improve confidence in PK/PD comparisons between the biosimilar and the reference product. High-quality bioanalytical data also strengthen regulatory submissions by demonstrating compliance with international guidelines. This contributes to a more reliable assessment of biosimilarity.
Before initiating a PK/PD study, companies should first establish strong analytical similarity between the biosimilar and its reference product. They should identify suitable pharmacodynamic biomarkers, validate all bioanalytical methods, and design statistically robust equivalence studies. Regulatory guidance from agencies such as the FDA, EMA, and Health Canada should also be carefully reviewed during study planning. Early scientific consultation and a well-defined development strategy can help avoid unnecessary delays and support a smoother regulatory approval process.
Reference
- Florian J, Sun Q, Schrieber SJ, White R, Shubow S, Johnson‐Williams BE, Sheikhy M, Harrison NR, Parker VJ, Wang YM, Strauss DG. Pharmacodynamic biomarkers for biosimilar development and approval: a workshop summary. Clinical Pharmacology & Therapeutics. 2023 May;113(5):1030-5.https://ascpt.onlinelibrary.wiley.com/doi/abs/10.1002/cpt.2795
- Florian J, Gershuny V, Sun Q, Schrieber SJ, Matta MK, Hazel A, Sheikhy M, Weaver JL, Hyland PL, Hsiao CH, Vegesna G. Considerations for use of Pharmacodynamic biomarkers to support biosimilar development–(III) a randomized trial with interferon Beta‐1a products. Clinical Pharmacology & Therapeutics. 2023 Feb;113(2):339-48.https://ascpt.onlinelibrary.wiley.com/doi/abs/10.1002/cpt.2784
- Niazi SK. Support for Removing Pharmacodynamic and Clinical Efficacy Testing of Biosimilars: A Critical Analysis. Clinical Pharmacology in Drug Development. 2023 Dec 1;12(12).https://search.ebscohost.com/login.aspx?direct=true&profile=ehost&scope=site&authtype=crawler&jrnl=2160763X&asa=N&AN=173973206&h=0DPOihfUVHJ9%2FUSFRegVuqVkUqq4Nh8rPqoPpHs%2BR4Re1go9GYf9nEqlr31t8SWMi8Y1uFrWDZa53NX5kVsgAQ%3D%3D&crl=c
- Sheikhy M, Schrieber SJ, Sun Q, Gershuny V, Matta MK, Bai JP, Du X, Vegesna G, Shah A, Prentice K, Nalepinski C. Considerations for use of pharmacodynamic biomarkers to support biosimilar development–(I) a randomized trial with PCSK9 inhibitors. Clinical Pharmacology & Therapeutics. 2023 Jan;113(1):71-9.https://ascpt.onlinelibrary.wiley.com/doi/abs/10.1002/cpt.2769
- Stebbing J, Mainwaring PN, Curigliano G, Pegram M, Latymer M, Bair AH, Rugo HS. Understanding the role of comparative clinical studies in the development of oncology biosimilars. Journal of Clinical Oncology. 2020 Apr 1;38(10):1070-80.https://link.springer.com/article/10.1208/s12248-018-0246-1

