 to Streamline Biosimilar Lot Release Testing")
Introduction:
Biosimilar development is analytically demanding. Regulatory agencies — Health Canada, the FDA, and the EMA alike — require manufacturers to demonstrate that every released lot meets a comprehensive set of predefined quality criteria. Historically, meeting these criteria has meant running a long list of orthogonal assays, including peptide mapping, glycan profiling for N-linked oligosaccharides, charge variant analysis, oxidation monitoring, and more. Each test consumes time, material, and skilled analyst hours. As biosimilar programs mature, advanced biosimilar characterization using mass spectrometry has become increasingly important for generating comprehensive analytical evidence while reducing workflow complexity.
The Multi-Attribute Method (MAM) for biosimilar lot release testing changes this equation fundamentally. By harnessing high-resolution liquid chromatography coupled with tandem mass spectrometry (LC-MS/MS), MAM simultaneously quantifies multiple Critical Quality Attributes (CQAs) in a single analytical run. This integrated approach supports robust biosimilar comparability studies while delivering the analytical depth required for regulatory submissions.
At ResolveMass Laboratories Inc., our analytical chemistry team has implemented MAM across multiple biosimilar development programs. As part of our comprehensive biosimilar characterization services, we integrate MAM with orthogonal analytical techniques to support comparability, regulatory filings, and commercial lot release. This case study documents one such implementation — a monoclonal antibody (mAb) biosimilar — describing the challenge, the MAM strategy applied, the analytical validation outcomes, and the regulatory positioning achieved.
Summary:
- The Multi-Attribute Method (MAM) is a mass spectrometry-based platform that consolidates multiple biosimilar lot release tests into a single analytical workflow.
- Traditional biosimilar lot release testing requires 8–12 separate assays; MAM reduces this to one unified LC-MS/MS method without sacrificing data quality.
- MAM simultaneously monitors glycosylation, deamidation, oxidation, sequence variants, and other critical quality attributes (CQAs) in one run.
- Implementing MAM for biosimilar lot release shortens cycle time, reduces sample consumption, and strengthens regulatory defensibility through integrated LC-MS/MS services for biosimilars.
- ResolveMass Laboratories Inc. has successfully applied MAM in biosimilar development programs, delivering faster turnaround and ICH-compliant analytical packages.
- This case study walks through a real-world MAM implementation for a monoclonal antibody (mAb) biosimilar, from method development through lot release readiness.
1: What Is the Multi-Attribute Method (MAM), and How Does It Work?
MAM is a quantitative LC-MS/MS-based peptide mapping method that simultaneously measures multiple protein quality attributes from a single sample digest. Instead of running separate assays for each attribute, MAM generates a comprehensive attribute profile—glycoforms, deamidation, oxidation, sequence variants, and more—from one injection. Advanced peptide mapping in biosimilars enables simultaneous sequence confirmation and modification analysis, while comprehensive peptide mapping and sequence analysis ensures structural integrity throughout development.
The core workflow involves four steps:
- Protein digestion — The biologic drug substance is digested with a site-specific protease (typically trypsin) under controlled conditions.
- LC-MS/MS analysis — Peptide fragments are separated by reversed-phase HPLC and detected by a high-resolution mass spectrometer (e.g., Q Exactive or Orbitrap platform).
- Attribute-level quantification — Software tools (such as BioPharma Finder or Byos) extract peak areas for modified and unmodified peptides and compute relative abundance for each attribute.
- New Peak Detection (NPD) — A surveillance function that automatically flags unexpected peaks not present in the reference standard, providing an additional layer of lot-to-lot consistency monitoring.
Key Quality Attributes Monitored by MAM
MAM simultaneously evaluates numerous structural and chemical modifications that influence biosimilar quality. Among these, glycosylation analysis of biosimilars is essential for understanding Fc-mediated biological activity, while monitoring post-translational modifications (PTMs) in biosimilars provides insight into product stability and manufacturing consistency (https://resolvemass.ca/glycosylation-analysis-of-biosimilars/, https://resolvemass.ca/post-translational-modifications-ptms-in-biosimilars/).
Likewise, disulfide bond mapping in biosimilars confirms correct protein folding and higher-order structural integrity, whereas advanced charge variant analysis in biosimilars helps characterize molecular heterogeneity and supports regulatory comparability assessments (https://resolvemass.ca/disulfide-bond-mapping-in-biosimilars/, https://resolvemass.ca/charge-variant-analysis-in-biosimilars-mass-spectrometry-approaches-for-heterogeneity/).
| Quality Attribute | Analytical Relevance | Regulatory Expectation |
|---|---|---|
| N-linked glycosylation (site-specific) | Efficacy, immunogenicity, Fc effector function | ICH Q6B; FDA Biosimilar Guidance |
| Deamidation (Asn → Asp) | Chemical degradation, potency loss | Stability and comparability data |
| Oxidation (Met, Trp) | Oxidative stress, shelf-life impact | Forced degradation profiling |
| Succinimide / isoAsp formation | Structural heterogeneity | CQA risk classification |
| C-terminal lysine clipping | Charge heterogeneity | Comparability to reference product |
| Sequence variants / misincorporation | Manufacturing process fidelity | ICH Q6B sequence confirmation |
| Disulfide scrambling | Higher-order structure integrity | Structural characterization |
Beyond these targeted CQAs, MAM workflows can also complement aggregation analysis in biosimilars, impurity profiling of biosimilars, and native mass spectrometry for biosimilars, enabling a broader analytical characterization strategy.
Because product quality directly impacts patient safety, analytical data generated through MAM also contributes to comprehensive immunogenicity assessment in biosimilar development, supporting regulatory confidence in product comparability.
2: The Case Study: MAM Implementation for a Monoclonal Antibody Biosimilar
Program Background
The biosimilar candidate was a full-length IgG1 monoclonal antibody targeting a validated oncology antigen, being developed to reference an innovator biologic approved in both the US and EU. The client required a complete analytical strategy for lot release, biosimilar comparability studies, and regulatory submission, capable of supporting both an Abbreviated Biologics License Application (aBLA) filing with the FDA and a biosimilar application with the EMA. Comprehensive biosimilar comparability studies form the foundation of regulatory approval by demonstrating that the proposed biosimilar closely matches the reference product across multiple analytical attributes.
To strengthen this analytical package, the development program also incorporated biosimilar characterization using mass spectrometry, which enables simultaneous evaluation of multiple structural and functional attributes with greater sensitivity than many conventional analytical methods.
The traditional testing panel proposed by the internal quality team included:
- Peptide mapping (sequence confirmation)
- Released glycan profiling (HILIC-FLD)
- Charge variant analysis (icIEF)
- Oxidation monitoring (RP-HPLC stressed samples)
- Deamidation profiling (LC-MS)
- Host cell protein (HCP) ELISA
- In-process purity (SEC-HPLC)
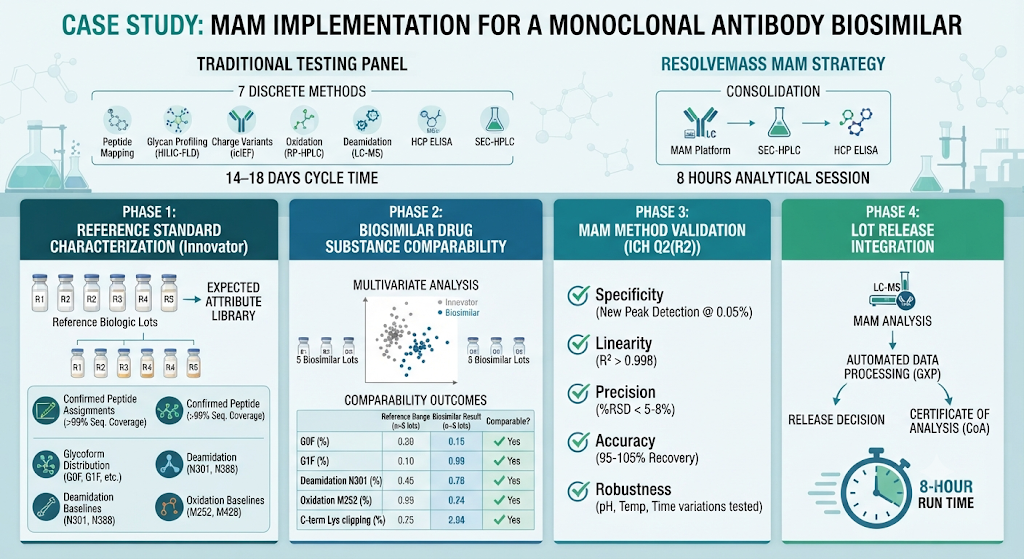
This represented seven discrete analytical methods, each requiring separate method validation, dedicated reference standards, and specialized analysts. The resulting workflow was resource-intensive, with cumulative cycle times ranging from 14–18 business days per lot.
Modern LC-MS platforms now make it possible to prove biosimilarity using LC-MS by integrating multiple analytical endpoints into a unified workflow. Combined with intact mass analysis for biosimilars, developers can simultaneously evaluate molecular weight, sequence integrity, post-translational modifications, and product heterogeneity while reducing analytical complexity.
The MAM Strategy Applied at ResolveMass
The ResolveMass analytical team proposed consolidating five of the seven assays into a single MAM platform, retaining SEC-HPLC for aggregation monitoring and HCP ELISA for process-related impurities (which fall outside MAM’s scope by design).
Phase 1 — Reference Standard Characterization
Before MAM could be used for lot release, a fully characterized reference standard panel was established. The innovator product (reference biologic, five independent lots) was digested and analyzed by MAM to build an expected attribute library. This library defined:
- Confirmed peptide assignments (sequence coverage > 99%)
- Expected glycoform distribution (G0F, G1F, G2F, Man5, afucosylated species)
- Baseline levels for deamidation at N301, N388, and heavy chain CDR2 sites
- Oxidation baselines at M252, M428 (Fc region)
- C-terminal lysine clipping profile
To further strengthen method robustness, the reference material also underwent forced degradation of biosimilars studies to establish degradation signatures under oxidative, thermal, and stress conditions. These studies helped define acceptable degradation boundaries and improve interpretation of stability-related CQAs during routine lot release.
Phase 2 — Biosimilar Drug Substance Characterization and Comparability
Five lots of the biosimilar drug substance were analyzed under the same MAM conditions. Statistical comparability was assessed using:
- Similarity Factor (f2) for glycoform profiles
- Quality range-setting based on the reference biologic data
- Multivariate analysis (PCA) across the full attribute vector
Comparability outcomes (selected attributes):
| Attribute | Reference Range (n=5 lots) | Biosimilar Result (n=5 lots) | Comparable? |
|---|---|---|---|
| G0F (%) | 42.1 ± 3.4 | 43.8 ± 2.9 | ✅ Yes |
| G1F (%) | 31.6 ± 2.1 | 30.4 ± 1.8 | ✅ Yes |
| Deamidation N301 (%) | 1.8 ± 0.4 | 2.0 ± 0.3 | ✅ Yes |
| Oxidation M252 (%) | 0.7 ± 0.2 | 0.8 ± 0.2 | ✅ Yes |
| C-term Lys clipping (%) | 88.4 ± 4.1 | 86.7 ± 3.6 | ✅ Yes |
The comprehensive analytical dataset generated through MAM aligned with the Totality of Evidence approach in biosimilar approval, where extensive analytical similarity forms the primary foundation for demonstrating biosimilarity before clinical studies are considered. High-quality analytical evidence significantly reduces uncertainty during regulatory review by confirming that any observed differences are not clinically meaningful.
Phase 3 — MAM Method Validation for Lot Release
Method validation was executed in accordance with ICH Q2(R2) and the FDA’s emerging guidance on MS-based lot release assays. Key validation parameters evaluated:
- Specificity — All targeted attributes resolved without interference from non-target peptides; New Peak Detection sensitivity verified at 0.05% relative abundance threshold.
- Linearity — Calibration curves for attribute-level quantification (R² > 0.998 across all monitored attributes).
- Precision (Repeatability and Intermediate Precision) — %RSD < 5% for all glycoforms; %RSD < 8% for low-abundance modification sites.
- Accuracy — Spiked recovery within 95–105% for all targeted attributes.
- Robustness — Digestion time (±30 min), column temperature (±2°C), and mobile phase pH (±0.1 unit) evaluated with no significant impact on attribute quantification.
Because MAM simultaneously evaluates sequence confirmation, PTMs, glycosylation, oxidation, deamidation, and other molecular attributes, it has become an increasingly valuable platform for analytical tests for biosimilar regulatory submission, providing a single integrated dataset that satisfies multiple regulatory expectations.
Phase 4 — Lot Release Integration
Upon validation, the MAM method was integrated into the lot release workflow. Each lot release run was executed within a single eight-hour analytical session, compared to the 14–18 days previously required for the full battery of separate assays.
3: How MAM Improves Biosimilar Lot Release: A Direct Comparison
The adoption of the Multi-Attribute Method (MAM) transforms biosimilar lot release from a collection of independent analytical tests into a single, integrated, high-resolution workflow. Rather than generating fragmented datasets from multiple orthogonal methods, MAM provides a unified analytical profile capable of simultaneously evaluating numerous Critical Quality Attributes (CQAs). This comprehensive approach not only accelerates lot release but also enhances data consistency, simplifies review, and strengthens regulatory confidence throughout the product lifecycle.
Because MAM combines peptide mapping with high-resolution LC-MS/MS, it enables developers to perform biosimilar characterization using mass spectrometry while supporting both routine quality control and analytical similarity assessments using the same validated platform.
Traditional Multi-Assay vs. MAM Workflow
| Parameter | Traditional Multi-Assay Panel | MAM-Integrated Workflow |
|---|---|---|
| Number of separate methods | 5–7 | 1 (MAM) + 2 orthogonal methods |
| Sample volume required per lot | ~15–25 mg protein | ~2–4 mg protein |
| Analyst time per lot release | 14–18 business days | 5–7 business days |
| Attributes monitored per run | 1–3 per method | 20+ simultaneously |
| New peak surveillance | Manual, assay-specific | Automated New Peak Detection |
| Data integration effort | Cross-method reconciliation | Single integrated report |
| Regulatory data package | Fragmented across reports | Consolidated attribute profile |
Compared with traditional workflows, MAM offers several operational and scientific advantages:
- Reduced analytical complexity by replacing multiple stand-alone methods with a single LC-MS/MS workflow.
- Lower sample consumption, preserving valuable development and reference materials.
- Improved laboratory efficiency, allowing analysts to evaluate numerous CQAs from a single injection.
- Consistent attribute monitoring, minimizing variability between independent analytical methods.
- Comprehensive molecular characterization, which supports regulatory comparability exercises and lifecycle management.
Furthermore, combining MAM with advanced LC-MS/MS services for biosimilars enables analytical laboratories to generate robust datasets suitable for both development and commercial quality control while reducing overall project timelines.
Another significant advantage is the ability to integrate biosimilar comparability studies directly into routine analytical workflows. Rather than repeating separate characterization studies for different development milestones, developers can leverage the same validated MAM platform throughout product development, technology transfer, process validation, and commercial manufacturing.
4: Regulatory Considerations for MAM as a Lot Release Method
Regulators including the FDA and EMA have shown increasing receptivity to MAM as part of biosimilar lot release and comparability packages, provided the method is rigorously validated. Several key regulatory considerations apply:
- ICH Q2(R2) Compliance — The 2023 revision to ICH Q2 explicitly addresses mass spectrometry-based methods, providing clearer validation expectations. ResolveMass designs all MAM validation packages to align with this updated guidance.
- FDA Biosimilar Action Plan — The FDA has encouraged the use of modern analytical tools including MAM in biosimilar comparability exercises. Sponsor interactions (Type B meetings) increasingly include MAM data as primary evidence for analytical similarity.
- EMA Biosimilar Guidelines — The EMA’s state-of-the-art guidance on physicochemical characterization embraces multiplexed attribute monitoring. MAM data has been accepted as supporting evidence in several approved European biosimilar applications.
Developers preparing submissions for both North American and European markets should understand the important differences between FDA vs. EMA biosimilar regulatory pathways, particularly regarding analytical expectations, comparability documentation, and regulatory review strategies. - Pharmacopoeial Alignment — While USP and EP do not yet have compendial MAM methods, the growing body of regulatory precedent from approved applications is establishing a de facto standard.
- New Peak Detection (NPD) as a Process Sentinel — Regulatory reviewers have recognized NPD as a meaningful process consistency tool. Documenting NPD thresholds and alert criteria in the method validation report strengthens the lot release package.
Supporting the Totality of Evidence
Analytical similarity remains the cornerstone of biosimilar approval.
Because MAM simultaneously evaluates sequence identity, glycosylation, PTMs, oxidation, deamidation, charge variants, and multiple additional CQAs, it aligns exceptionally well with the Totality of Evidence approach in biosimilar approval, where robust analytical characterization significantly reduces residual uncertainty before nonclinical and clinical studies.
Comprehensive analytical characterization also helps reduce the likelihood of biosimilar comparability failure by identifying meaningful differences early in development and supporting scientifically justified acceptance criteria before regulatory submission.
Similarly, developers that implement extensive analytical characterization throughout development are better positioned to avoid common issues that explain why biosimilars fail regulatory approval, including insufficient analytical similarity, poorly justified CQAs, inadequate comparability packages, and incomplete validation documentation.
Finally, MAM-generated datasets can be incorporated into broader analytical tests for biosimilar regulatory submission, providing a unified evidence package that supports regulatory review while reducing reliance on numerous independent analytical reports.
5: Key Advantages of MAM for Biosimilar Development Programs
Beyond lot release efficiency, MAM delivers strategic advantages across the biosimilar development timeline:
- Accelerated comparability studies — Attribute-level comparability data generated from MAM can populate multiple sections of the aBLA/BLA comparability report simultaneously.
One of the greatest advantages of MAM is its ability to streamline biosimilar comparability studies. Rather than generating separate datasets from multiple orthogonal methods, MAM simultaneously evaluates numerous CQAs in a single analytical workflow. This integrated approach simplifies analytical similarity assessments and produces data that can populate multiple sections of the regulatory dossier while supporting the Totality of Evidence approach in biosimilar approval. - Earlier CQA identification — By monitoring 20+ attributes from early-stage batches, development teams can identify critical quality attributes before clinical manufacturing.
- Reduced reference standard consumption — Fewer separate assays means less depletion of limited innovator product lots used as reference standards.
- Lifecycle management efficiency — Post-approval changes (manufacturing site, cell line optimization, process changes) can be assessed rapidly using the same validated MAM platform, maintaining analytical continuity.
- Integrated stability profiling — MAM tracks degradation pathways (deamidation, oxidation) with high sensitivity, enabling more informative stability studies under ICH Q1A conditions.
6: Why ResolveMass Laboratories Is the Right MAM Partner
ResolveMass Laboratories Inc. is a Canadian Generic Pharmaceutical CDMO and Contract Research Organization (CRO) specializing in advanced analytical characterization for biologics, biosimilars, peptides, and complex pharmaceuticals. Our integrated scientific expertise combines regulatory knowledge with state-of-the-art analytical technologies to support biosimilar developers from early characterization through commercial lot release. Learn more about our Generic Pharmaceutical CDMO capabilities:
- High-resolution LC-MS/MS instrumentation operating in data-independent acquisition (DIA) and targeted MS² modes, optimized for biotherapeutic peptide mapping.
- Dedicated biopharmaceutical analytical scientists with hands-on experience implementing MAM for IgG1, IgG2, IgG4, Fc-fusion, and bispecific antibody formats.
- Regulatory-ready documentation — Every MAM engagement at ResolveMass is designed from the outset to produce ICH-compliant validation reports, comparability summaries, and data packages suitable for aBLA/BLA/NDS submissions.
- Integrated analytical platform — MAM at ResolveMass is delivered as part of a broader characterization package that includes SEC-HPLC, HCP, host cell DNA, potency, and identity testing — enabling a single-source solution for biosimilar lot release.
- Transparent project management — Clients receive real-time project updates, raw data access, and dedicated analytical support throughout the program lifecycle.
Conclusion:
The Multi-Attribute Method (MAM) for biosimilar lot release testing is no longer an emerging technology — it is a proven, validated analytical platform that is reshaping the efficiency and regulatory defensibility of biosimilar development. As this case study demonstrates, MAM can consolidate five or more individual assays into a single high-information workflow, cutting lot release cycle times by over 60%, reducing sample consumption dramatically, and generating richer comparability data than any single traditional method.
For biosimilar developers operating in an increasingly competitive and timeline-driven market, adopting MAM is both a scientific and strategic decision. The earlier MAM is implemented in the development program — ideally from Phase 1 characterization through IND-enabling studies — the greater the cumulative benefit across the product lifecycle.
MAM is particularly valuable for emerging peptide and protein therapeutics. ResolveMass supports GLP-1 biosimilar characterization, insulin biosimilar characterization, and peptide biosimilar characterization using LC-MS, helping developers generate the comprehensive analytical data required for successful biosimilar submissions.
- GLP-1 Biosimilar Characterization.
- Insulin Biosimilar Characterization.
- Peptide Biosimilar Characterization Using LC-MS.
For peptide therapeutics and GLP-1 products, developers must also consider evolving GLP-1 peptide characterization regulatory requirements, GLP-1 peptide mapping regulatory requirements, and the important analytical distinction between Peptide Sameness vs. Biosimilar Comparability when designing regulatory strategies.
- GLP-1 Peptide Characterization Regulatory Requirements.
- GLP-1 Peptide Mapping Regulatory Requirements.
- Peptide Sameness vs. Biosimilar Comparability.
For sustained-release peptide formulations, complementary Peptide–PLGA Interaction Analysis provides additional insight into formulation performance and product stability.
ResolveMass Laboratories Inc. brings the instrumentation, validated methods, regulatory expertise, and project management experience to implement Multi-Attribute Method (MAM) for biosimilar lot release efficiently and compliantly. Whether you are initiating a new biosimilar program, preparing for a comparability exercise, or seeking to modernize your existing lot release panel, our team is ready to support you.
Frequently Asked Questions:
MAM improves biosimilar lot release testing by combining multiple assays into one streamlined LC-MS workflow. This reduces overall testing time and operational complexity. It provides more detailed and accurate structural information compared to conventional methods. MAM helps detect subtle changes in protein structure that may be missed by traditional assays. It also improves batch-to-batch consistency evaluation. The method supports faster decision-making for product release. Overall, it enhances efficiency and analytical confidence in biosimilar manufacturing.
MAM is better because it provides simultaneous multi-attribute monitoring instead of single-parameter testing. Traditional methods require separate assays for each quality attribute, which increases time and variability. MAM uses high-resolution LC-MS to deliver precise, site-specific insights. It reduces redundancy in testing workflows and improves data integration. The method also enhances sensitivity to detect minor structural changes. It supports a more holistic understanding of biosimilar quality. This makes it a more advanced and efficient analytical strategy.
Yes, MAM is increasingly recognized by regulatory agencies such as the FDA and EMA when properly validated. It aligns with Quality by Design (QbD) principles and modern biologics development expectations. Regulators encourage its use for detailed product characterization and comparability studies. However, full method validation is required before routine application in lot release. Companies must demonstrate accuracy, precision, and robustness. Regulatory acceptance is growing as more industry case studies emerge. It is considered a promising tool for advanced biologics analytics.
MAM can detect a wide range of critical quality attributes in biosimilars. These include post-translational modifications such as oxidation, deamidation, and glycation. It also identifies amino acid sequence variants and structural changes. MAM can monitor product-related impurities and degradation products. It provides site-specific mapping of modifications within the protein structure. This helps ensure product consistency and safety. The method gives a comprehensive quality profile in a single run.
MAM can replace many traditional assays but not always all of them. It significantly reduces the need for multiple orthogonal methods in lot release testing. However, some functional bioassays may still be required depending on regulatory expectations. MAM is mainly used for structural and chemical quality attribute analysis. It complements rather than completely eliminates certain biological activity tests. The extent of replacement depends on product type and validation strategy. It is best used as part of an integrated analytical approach.
Implementing MAM requires advanced LC-MS instrumentation and skilled analytical expertise. The initial setup cost can be high compared to traditional methods. Data processing and interpretation are more complex due to high-resolution outputs. Method development and validation can be time-intensive. Regulatory acceptance requires strong documentation and robust validation studies. Training and standardization are also important challenges. Despite this, long-term benefits often outweigh the initial complexity.
MAM supports biosimilar development by enabling detailed comparability studies with reference products. It helps identify structural differences early in development. This reduces risks during later manufacturing stages. MAM improves understanding of critical quality attributes across batches. It supports Quality by Design (QbD)-based development strategies. The method enhances decision-making in formulation and process optimization. Overall, it accelerates biosimilar development timelines.
Reference
- Millán‐Martín S, Jakes C, Carillo S, Bones J. Multi‐Attribute Method (MAM) Analytical Workflow for Biotherapeutic Protein Characterization from Process Development to QC. Current Protocols. 2023 Nov;3(11):e927.https://currentprotocols.onlinelibrary.wiley.com/doi/abs/10.1002/cpz1.927
- Millán-Martín S, Jakes C, Carillo S, Rogers R, Ren D, Bones J. Comprehensive multi-attribute method workflow for biotherapeutic characterization and current good manufacturing practices testing. Nature Protocols. 2023 Apr;18(4):1056-89.https://www.nature.com/articles/s41596-022-00785-5
- Rogers RS, Abernathy M, Richardson DD, Rouse JC, Sperry JB, Swann P, Wypych J, Yu C, Zang L, Deshpande R. A view on the importance of “multi-attribute method” for measuring purity of biopharmaceuticals and improving overall control strategy. The AAPS journal. 2017 Nov 30;20(1):7.https://link.springer.com/article/10.1208/s12248-017-0168-3
- Yang F, Zhang J, Buettner A, Vosika E, Sadek M, Hao Z, Reusch D, Koenig M, Chan W, Bathke A, Pallat H. Mass spectrometry-based multi-attribute method in protein therapeutics product quality monitoring and quality control. InMAbs 2023 Dec 31 (Vol. 15, No. 1, p. 2197668). Taylor & Francis.https://www.tandfonline.com/doi/abs/10.1080/19420862.2023.2197668
- Millán-Martín S, Jakes C, Carillo S, Gallagher L, Scheffler K, Broster K, Bones J. Multi-attribute method (MAM): an emerging analytical workflow for biopharmaceutical characterization, batch release and cGMP purity testing at the peptide and intact protein level. Critical reviews in analytical chemistry. 2024 Nov 16;54(8):3234-51.https://www.tandfonline.com/doi/abs/10.1080/10408347.2023.2238058

